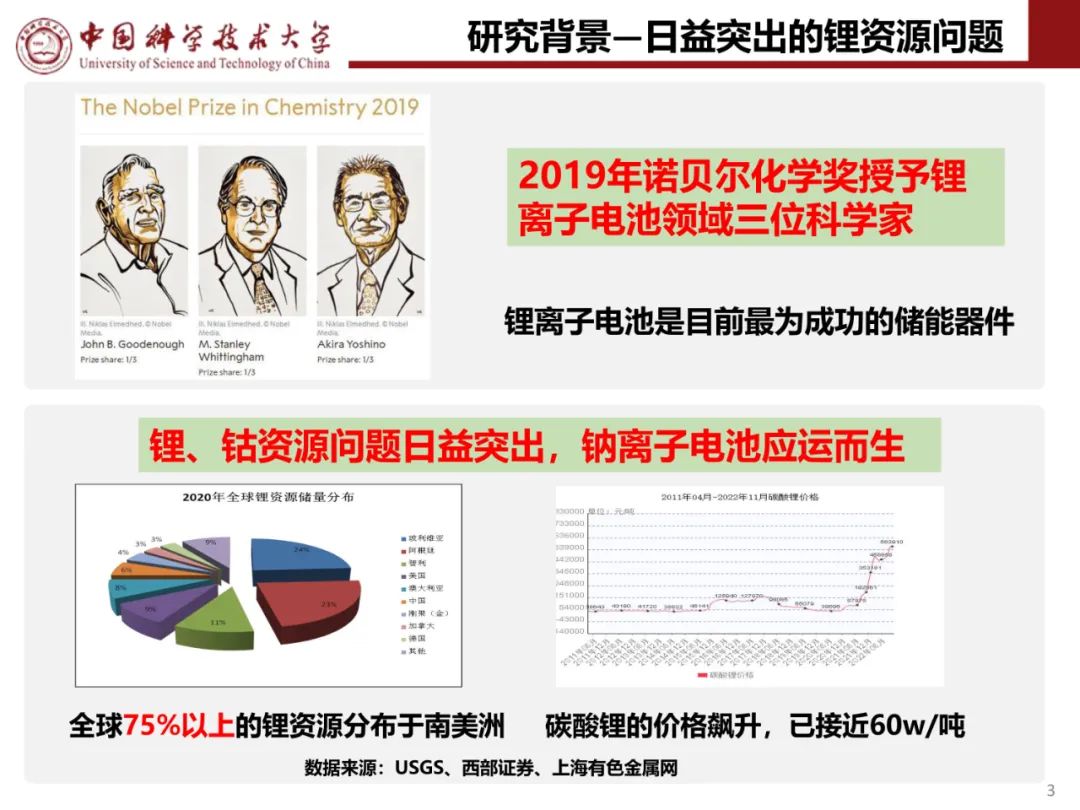
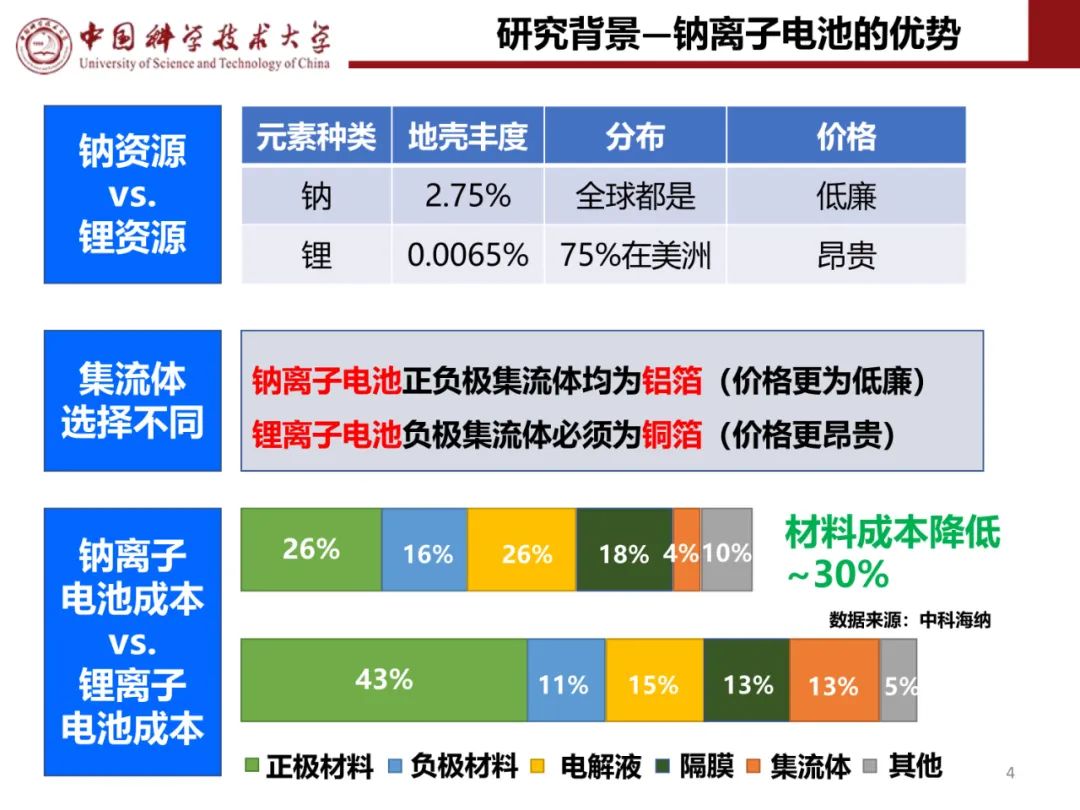
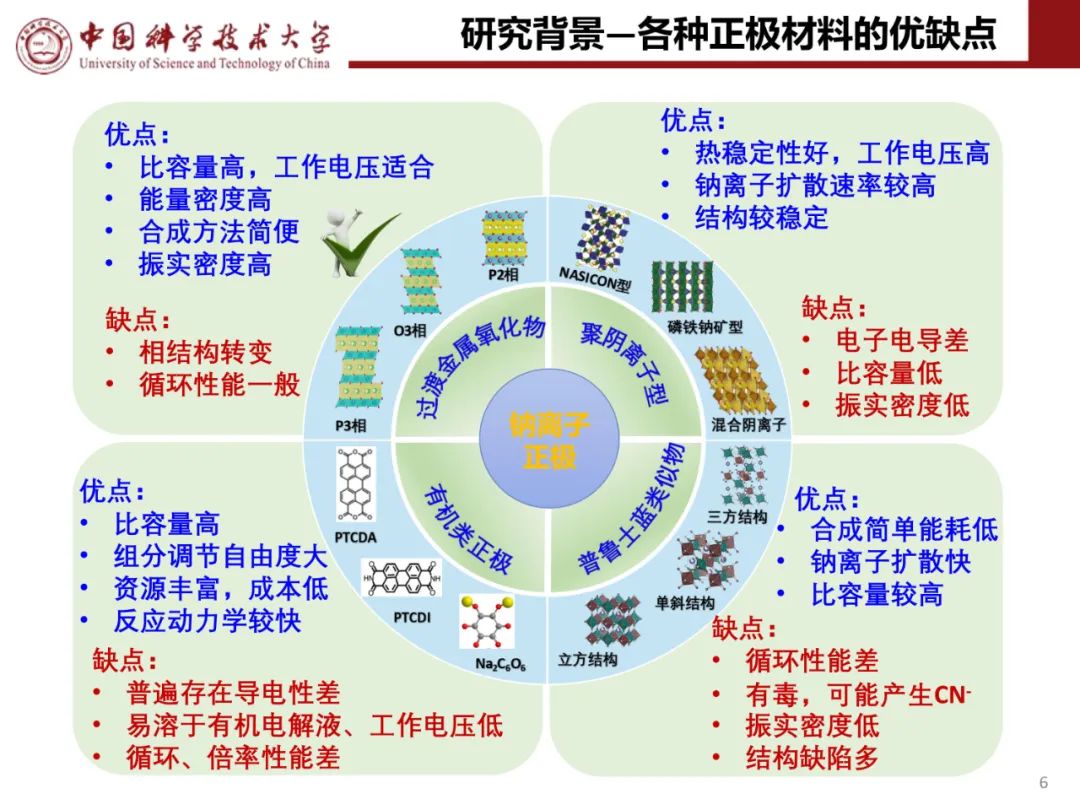
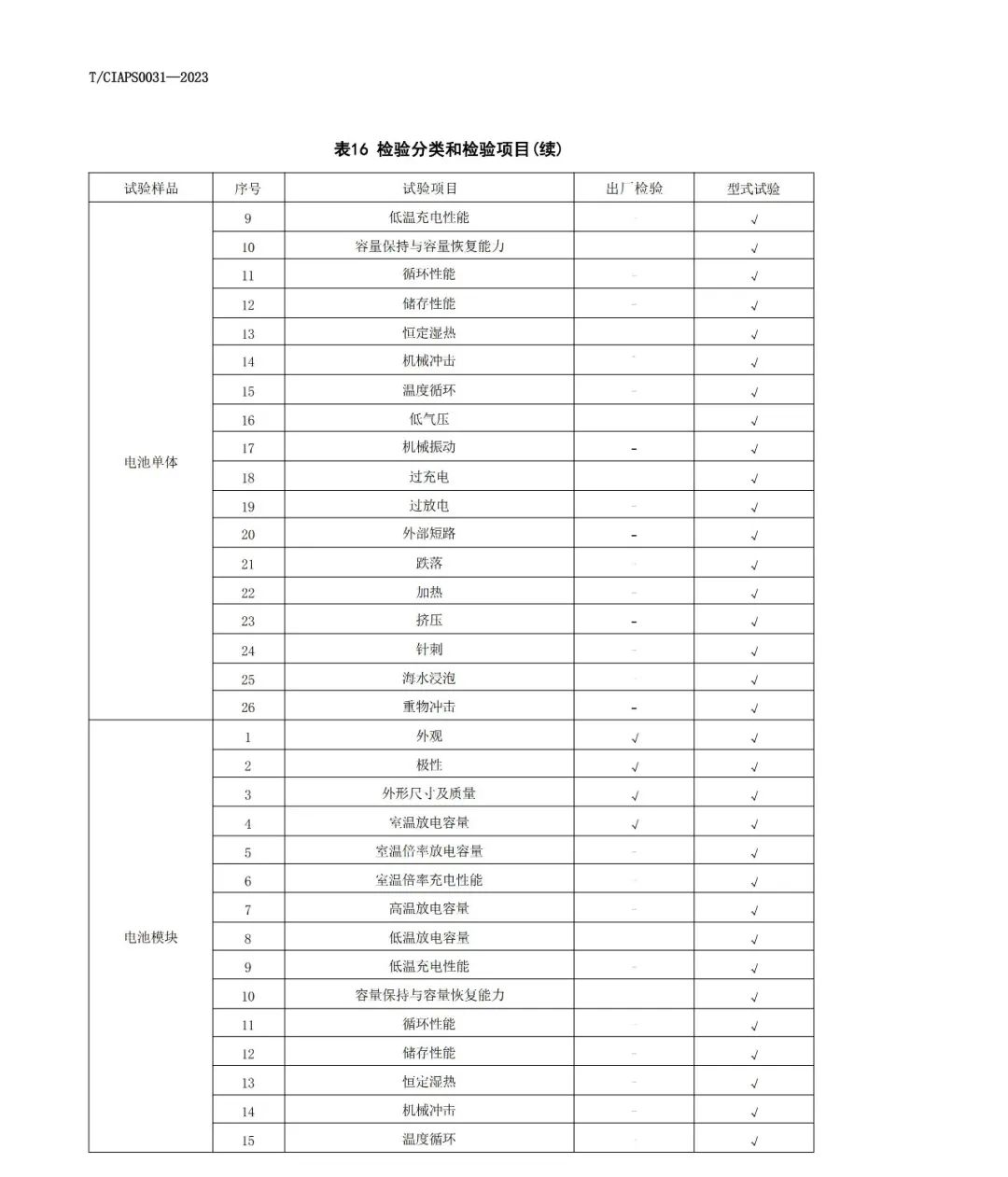
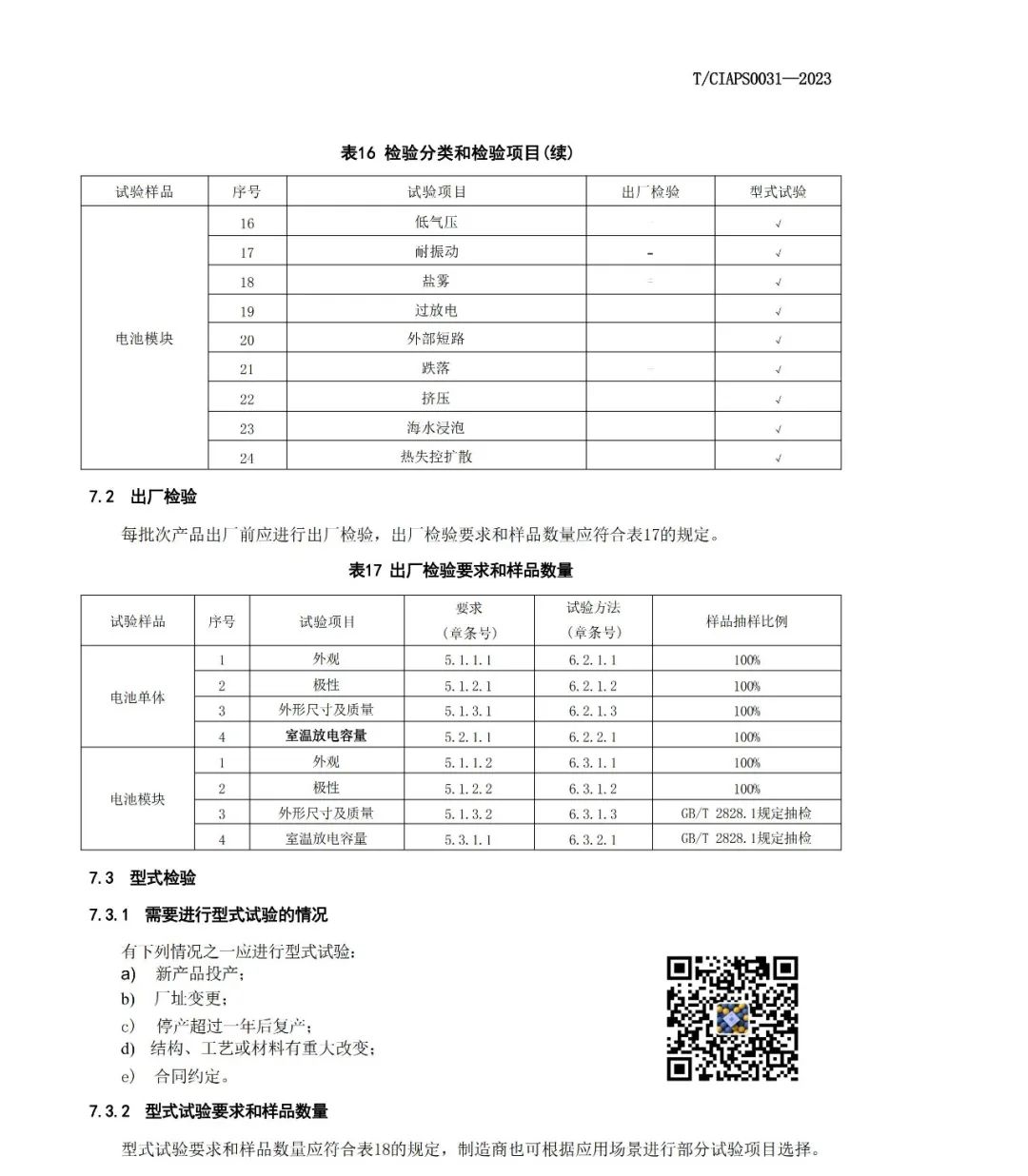
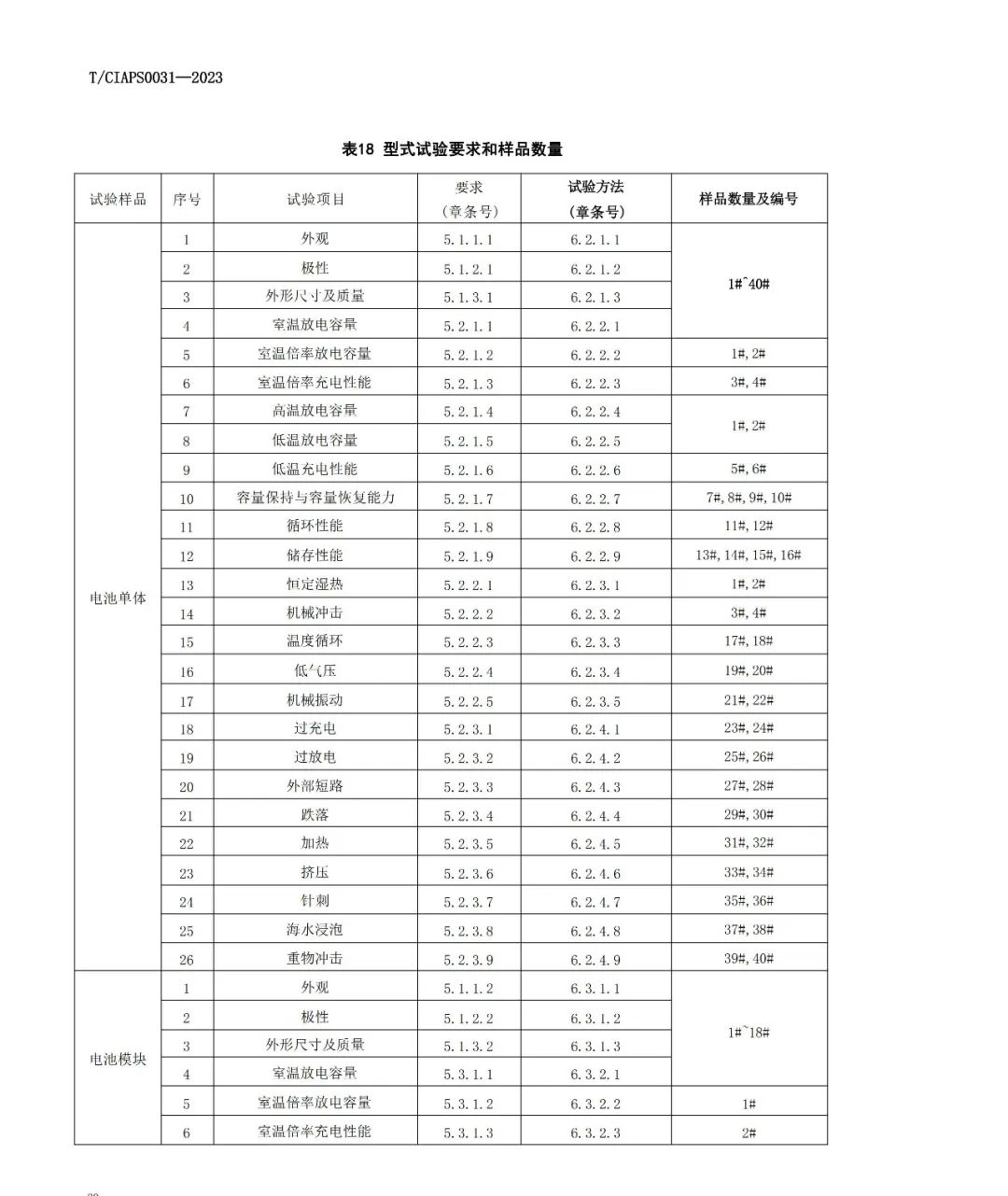
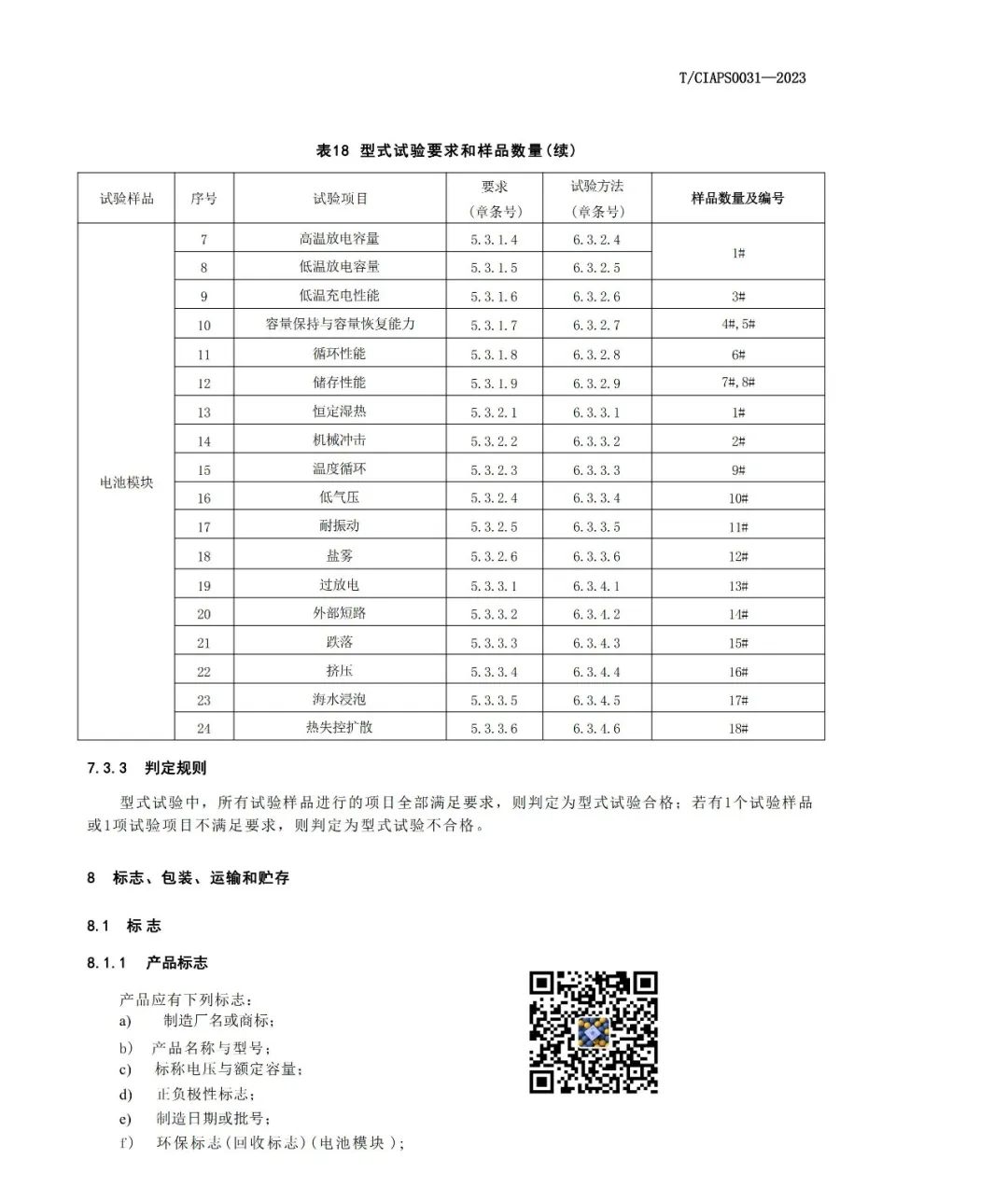
锂离子电池作为一种高性能的二次绿色电池,具有高电压、高能量密度(包括体积能量、质量比能量)、低的自放电率、宽的使用温度范围、长的循环寿命、环保、无记忆效应以及可以大电流充放电等优点;锂离子电池性能的改善,很大程度上决定于电极材料性能的改善,尤其是正极材料,目前研究最广泛的正极材料有LiCoO2、LiNiO2以及LiMn2O4等,但由于钴有毒且资源有限,镍酸锂制备困难,锰酸锂的循环性能和高温性能差等因素,制约了它们的应用和发展;因此开发新型高能廉价的正极材料对锂离子电池的发展至关重要;磷酸铁锂是一种新型的锂离子电池正极材料,具有高能量密度、长循环寿命、安全性好等优点,是首选的新一代绿色正极材料,特别是作为动力锂离子电池材料;磷酸铁锂的发现引起了国内外电化学界不少研究人员的关注,近几年,随着锂电池的越来越广的应用,对LiFePO4的研究越来越多;因此在电动汽车、储能系统等领域得到广泛应用。
1、磷酸铁锂的结构和性能:磷酸铁锂(LiFePO4)具有橄榄石结构,为稍微扭曲的六方密堆积,其空间群是Pmnb型,晶型结构图1所示。
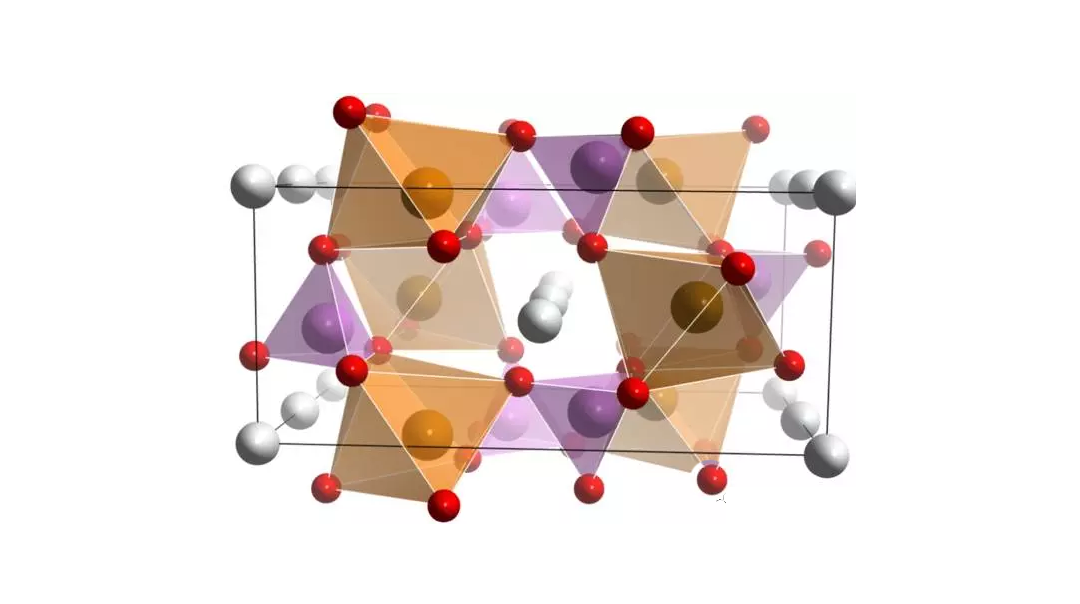
图 1 磷酸铁锂的空间结构图
LiFePO4由FeO6八面体和PO4四面体构成空间骨架,P占据四面体位置,而Fe和Li则填充在八面体空隙中,其中Fe占据共角的八面体位置,Li则占据共边的八面体位置。晶格一个FeO6八面体与两个FeO6八面体和一个PO4四面体共边,而PO4四面体则与一个FeO6八面体和两个LiO6八面体共边。由于近乎六方堆积的氧原子的紧密排列,使得锂离子只能在二维平面上进行脱嵌,也因此具有了相对较高的理论密度(3.6g/cm3)。在此结构中,Fe2+/Fe3+相对金属锂的电压为3.4V,材料的理论比容量为170mA·h/g。在材料中形成较强的P-O-M共价键,极大地稳定了材料的晶体结构,从而导致材料具有很高的热稳定性;科学家对LiFePO4的电化学性能做了详细的分析,图2是LiFePO4的循环载荷伏安图,在C-V图中形成两个峰,在阳极扫描时Li+从LixFePO4结构中脱出,在3.52V形成氧化峰;当在4.0~3.0扫描时Li+嵌入到LixFePO4结构中,相应的在3.32V形成还原峰;C-V曲线中的氧化还原峰表明在LiFePO4电极上发生着可逆的锂离子嵌脱反应。

图2磷酸铁锂的循环载荷伏安图
2、磷酸铁锂的制备方法及研究:LiFePO4正极材料的性能在一定程度上取决于材料的形态、颗粒的尺寸以及原子排列,因此制备方法尤为重要,目前主要有固相法和液相法,其中固相法包括高温固相反应法、碳热还原法、微波合成法和脉冲激光沉积法;液相法包括溶胶凝胶法、水热合成法、沉淀法以及溶剂热合成法等。
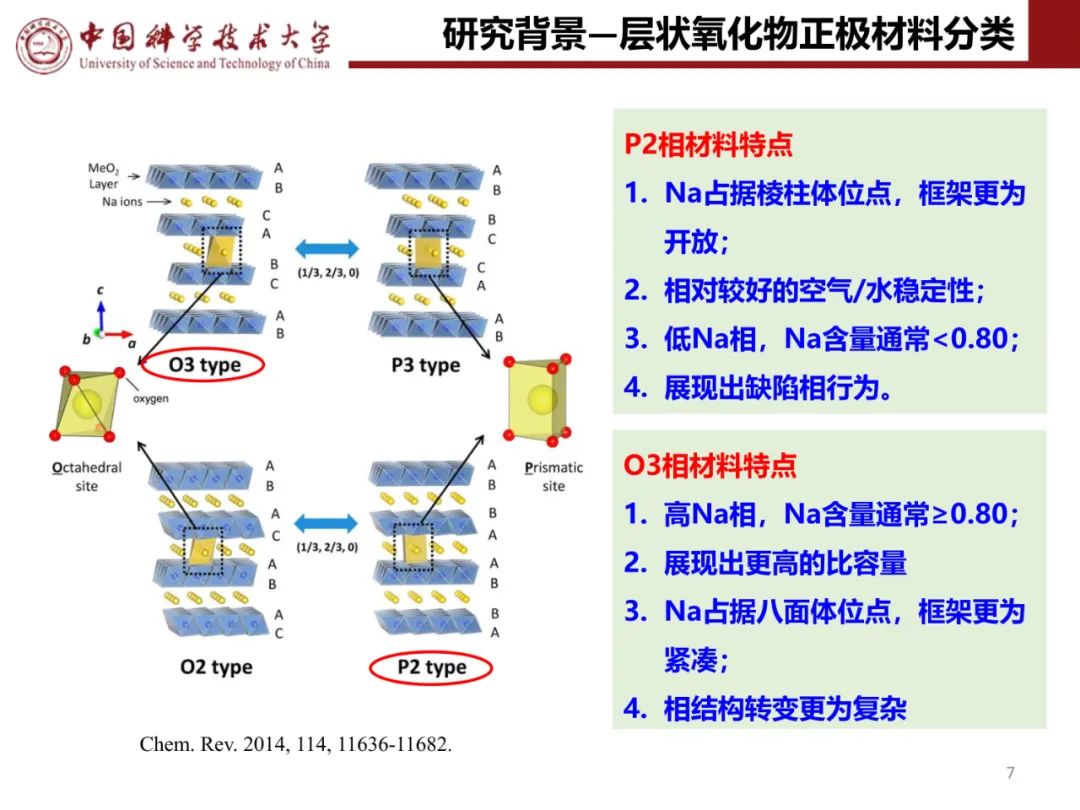
1)磷酸铁锂材料的固相法制备方法主要有:
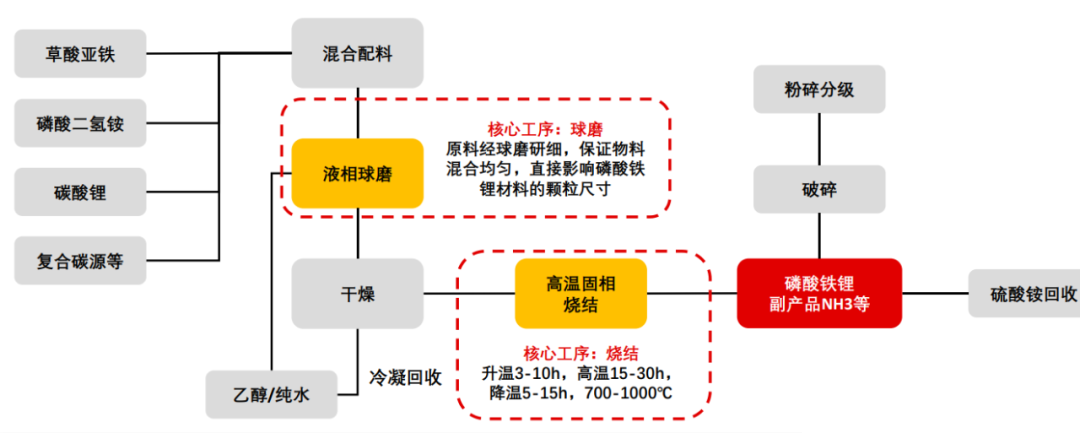
(1)高温固相法:J.Barker等就磷酸盐正极材料申请了专利,主要采用固相合成法;以碳酸锂、氢氧化锂等为锂源,草酸亚铁、乙二酸亚铁,氧化铁和磷酸铁等为铁源,磷酸根主要来源于磷酸二氢铵等。*典型的工艺流程为高温固相法:将原料球磨干燥后,在马弗炉或管式炉内于惰性或者还原气氛中,以一定的升温加速加热到某一温度,反应一段时间后冷却。
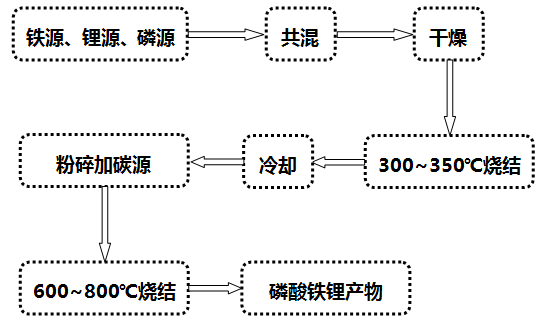

*该种合成方法容易在形成橄榄石结构中发生Fe错位现象,影响电化学性能,且水热法需要耐高温高压设备,工业化生产的困难要大一些。据称Phostech的P粉末便采用该类工艺生产。
*碳热还原法也是固相法中的一种,是比较容易工业化的合成方法,以廉价的三价铁作为铁源,通过高温还原的方法制备覆碳的LiFePO4复合材料。多数研究以磷酸二氢锂 (LiH2PO4)、三氧化二铁(Fe2O3)或四氧化三铁、蔗糖为原料,均匀混合后,在高温和氩气或氮气保护下焙烧,碳将三价铁还原为二价铁,也就是通过碳热还原法合成磷酸铁锂。
*Mich等以FePO4·4H2O和LiOH·H2O为原料,聚丙烯为还原剂,在氮气氛下500~800℃处理10h,合成的覆碳材料在0.1C及0.5C倍率下首次放电比容量分别为160mA·h/g和146.5mA·h/g。
*张宝等采用改进的碳热还原法,即以FeSO4·7H2O和NH4H2PO4为原料,采用液相沉淀法制备FePO4前驱体,然后将前驱体、Li2CO3及导电碳黑混合均匀,在Ar气的保护下分别在500、560、600、700和800℃下煅烧12h,合成LiFePO4;研究表明,560、600、700和800℃合成的样品均为LiFePO4/C,LiFePO4颗粒粒径随合成温度的升高而逐渐增大;560℃样品在放电倍率为0.1C时的首次放电比容量为151mA·h/g(0.1C),而当放电倍率达到1C时,放电比容量为129mA·h/g,且具有良好的循环性能。
*碳热还原法优点:采用碳热还原法解决了原料价格昂贵的缺点,能够广泛的应用于工业生产。还解决了在原料混合加工过程中可能引发的氧化反应,使合成过程更为合理,同时改善了材料的导电性。
*碳热还原法缺点:反应时间相对过长,温度难以控制,产物一致性要求的控制条件更为苛刻,难以适应工业化生产。
*高温固相法的优点是工艺简单、易实现产业化,但产物粒径不易控制、分布不均匀,形貌也不规则,并且在合成过程中需要使用惰性气体保护。
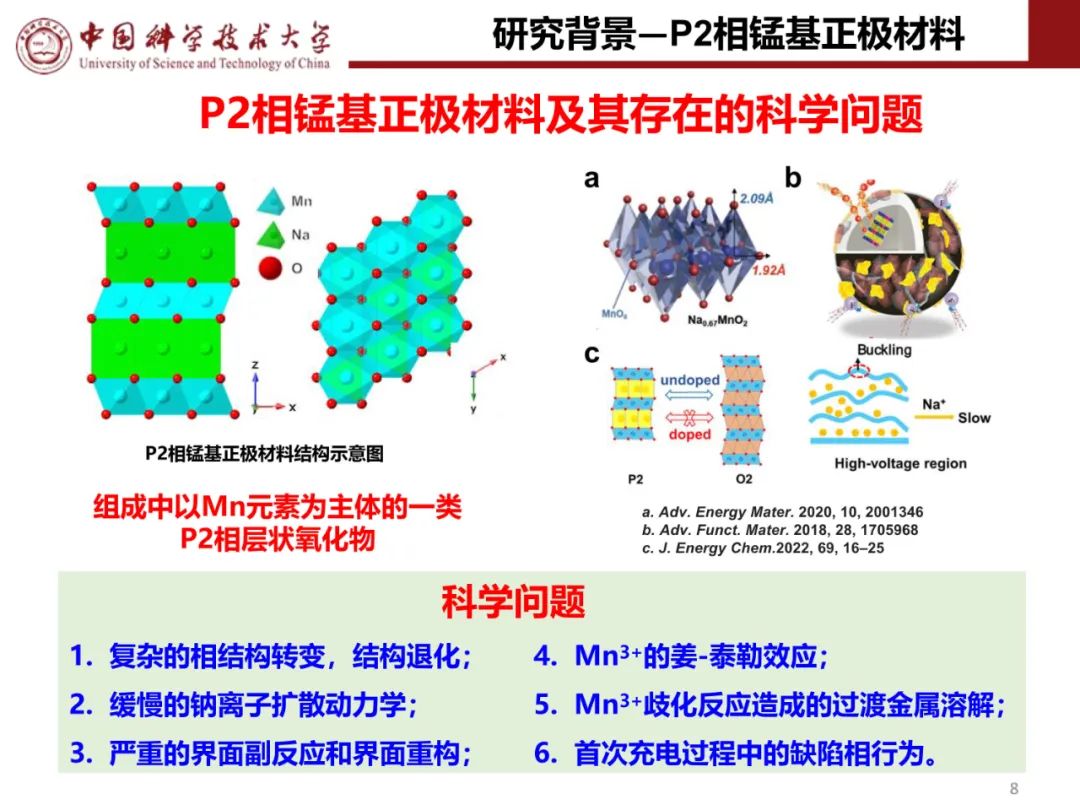
(2)碳热还原法:这种方法是高温固相法的改进,直接以铁的高价氧化物如FeO、LiHPO和碳粉为原料,以化学计量比混合,在箱式烧结炉氩气气氛中于700℃烧结一段时间,之后自然冷却到室温。
*采用该方法做成的实验电池首次充放电容量为151mAh/g。该方法目前有少数几家企业在应用,由于该法的生产过程较为简单可控,且采用一次烧结,所以它为LiFePO走向工业化提供了另一条途径;但该法制备的材料较传统的高温固相法容量表现和倍率性能方面偏低。
*HPO和FeCL合成FePO.2HO,然后与CHCOOLi通过水热法合成LiFePO。与高温固相法比较,水热法合成的温度较低,约150度~200度,反应时间也仅为固相反应的1/5左右,并且可以直接得到磷酸铁锂,不需要惰性气体,产物晶粒较小、物相均一等优点,尤其适合于高倍率放电领域。

(3)脉冲激光沉积法:Iriyama等首先使用固相合成方法制备出LiFePO4,然后将材料压片后在A r中800℃煅烧24h,使用常规的脉冲激光沉积系统得到薄层的LiFePO4,具有良好的循环性能,循环100周后容量保持初始容量的90%;Sauvage等通过研究不同厚度LiFePO4薄膜的电化学性能,他们发现离子电导率是限制薄膜电极的主要因素。该方法是一种制备薄膜电极的方法,但是需特殊设备。

(4)液相共沉淀法:该法原料分散均匀,前躯体可以在低温条件下合成。将LiOH加入到(NH)Fe(SO).6HO与HPO的混合溶液中,得到共沉淀物,过滤洗涤后,在惰性气氛下进行热处理,可以得到LiFePO;产物表现出较好的循环稳定性。日本企业采用这一技术路线,但因专利问题目前尚未大规模应用。

(5)雾化热解法:雾化热解法主要用来合成前躯体。将原料和分散剂在高速搅拌下形成浆状物,然后在雾化干燥设备内进行热解反应,得到前躯体,灼烧后得到产品。

(6)氧化-还原法:该法能得到电化学优良的纳米级的磷酸铁锂粉体,但其工艺很复杂,不能大量生产,只适合实验室研究。

(7)此外,还有乳化干燥法、微波烧结法及溶胶-凝胶法等。



2)目前国内外已经能实现磷酸铁锂电池量产的合成方法:均是高温固相法,高温固相法又分传统的(以天津斯特兰、湖南瑞翔、北大先行等为代表,以草酸亚铁做为铁源)和改进的(以美国Valence、苏州恒正为代表,以三价铁物质做为铁源,该法也称碳热还原法)两种。对碳热还原法来讲,选取的铁源主要有两种,一种是Valence的氧化铁红路线,还有一种是清华大学(已成立北京锂先锋科技)以及武汉大学(已转让浙江振华新能源)的技术,选用磷酸铁做为铁源,该法制程工艺较为简单,其最大优点是避开了其它合成方法中使用磷酸二氢铵为原料,产生大量氨气污染环境的问题,但对磷酸铁原料要求较高;目前清华大学的一个研究小组通过控制沉淀条件合成了一种粒度可控,碳掺杂的磷酸铁前驱体,但该法合成难度较高,在工业放大过程中面临一些问题;目前有些厂家选用磷酸二氢锂做为生产磷酸铁锂的原材料,同样可以避免反应过程的污染问题,这个在氧化铁红路线上有所体现;这条路线和磷酸铁加碳酸锂的路线均不产生污染。
3)磷酸铁锂材料的液相法制备方法主要有:
(1)水热合成法:水热法是指在高温高压下,在水或者蒸汽等流体中进行的有关化学反应的总称。水热技术有两个特点:一是其相对低的温度,二是在封闭容器中进行,避免了组分挥发。
*水热合成法属于湿法范畴,它是以可溶性亚铁盐、锂盐和磷酸为原料,在水热条件下直接合成 LiFePO4,由于氧气在水热体系中的溶解度很小,水热体系LiFePO4的合成提供了优良的惰性环境。
*张俊玲以量LiOH·H2O、FeSO4·7H2O、H3PO4为原料,加入少量的表面活性剂(预计产物量的2wt%),置于密封的釜体中升温至180℃保温4h,然后以预定降温速度进行冷却降温至100℃以下,过滤、洗涤,样品于120℃下真空干燥2h,将所得粉体与15%葡萄糖混合,放入管式炉,N2保护下600℃保温2h,得碳包裹的LiFePO4/C复合材料。结果表明,在30℃的环境温度下,材料0.2C、1C和5C首次充放电比容量分别为157、152和136mA·h/g,经过35次5C倍率充放电循环后,比容量无衰减。
*水热合成法优点:水热法可以在液相中制备超微细颗粒,原料可以在分子级混合。具有物相均匀、粉体粒径小以及操作简便等优点,且具有易量产、产品批量稳定性好、原料价廉易得的优点。同时生产过程中不需要惰性气氛。
*采用水热合成法可以得到晶形良好的LiMPO4,但是为了加入导电碳,在水溶液中加入聚乙二醇,再借由热处理过程转变为碳。
*水热合成法缺点:水热合成法制备的产物结构中常常存在着铁的错位,生成了亚稳态FePO4,影响了产物的化学及电化学性能。同时也存在粒径不均匀、物相不纯净、设备投资大(耐高温高压反应器的设计制造难度大,造价也高)或工艺较复杂的缺点。
(2)溶剂热合成法:溶剂热合成法与水热合成法相对应是用有机溶剂或水和有机溶剂混合物代替水做介质,采用类似水热合成的原理。
*甘晖等以溶剂热方法首次合成了橄榄石相的磷酸亚铁锂,并以水热法为参照;结果表明,使用溶剂热方法合成的磷酸亚铁锂是球形或多面体状、橄榄石相,随着反应时间的增加,颗粒逐渐长大;而使用水热方法合成磷酸亚铁锂时,颗粒由纤维状逐渐成长为菱形。
*周文彩认为,这两种方法产物的形状差异可能是由于溶剂热反应体系中较高的压力抑制了纤维态晶体的产生。
(3)雾化分解法:雾化分解法是一种获得小尺寸、规则形态的材料的有效方法,即将载气流通过超声喷雾的方法通入到高温(450~650℃)反应器中;将Li2CO3、FeC2O4和NH4H2PO4溶解在酸中,再加入一定量的蔗糖就可以得到前躯体,蔗糖作为碳源提供还原性气氛,空气作为载气流;此外超声喷雾分解可以制备掺杂金属的LiFePO4/C复合材料。
*这种方法制备的粉体呈球形但是结晶程度低,所以在600~900℃弱还原性气氛中的后期退火是必须环节。
*然而,在煅烧过程中的规则球形会有所改变。
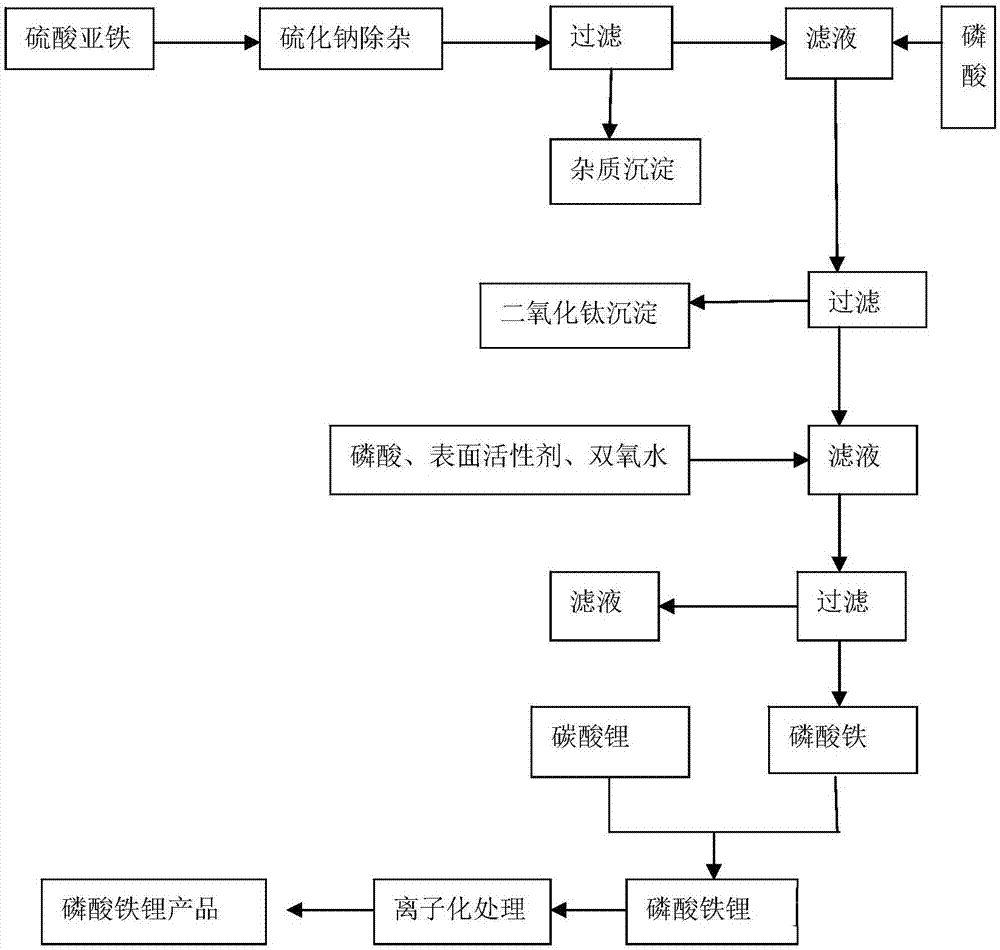
3、磷酸铁锂的生产工艺流程。
1)原料准备:磷酸铁锂的主要原料为氢氧化铁、磷酸、碳酸锂等。这些原料需要经过粉碎、筛分等处理,以保证其粒度均匀。
2)混合:将粉碎后的原料按一定比例混合,形成均匀的混合物。混合过程需要控制温度、湿度等参数,以确保混合物的质量。
3)烧结:将混合物放入烧结炉中进行烧结。烧结过程中需要控制温度、气氛等参数,以确保烧结物的结晶度和纯度。烧结后的产物为磷酸铁锂粉末。
4)磨碎:将磷酸铁锂粉末进行磨碎,以得到所需的粒度和形状。
5)包覆:将磷酸铁锂粉末与导电剂、粘结剂等混合,形成电极浆料。然后将电极浆料涂覆在铝箔或铜箔上,形成电极片。
6)组装:将电极片与负极片、隔膜等组装成电池,然后进行充电和放电测试,以确保电池的性能符合要求。

以上就是磷酸铁锂的生产工艺流程。需要注意的是,生产过程中需要严格控制各项参数,以确保产品的质量和性能;同时还需要采取环保措施,减少对环境的影响。
4、磷酸铁锂生产技术工艺经济比较:依据工艺路线的不同,生产磷酸铁锂正极材料的主要原材料有草酸亚铁、氧化铁红、磷酸铁、磷酸二氢锂和磷酸二氢铵等。

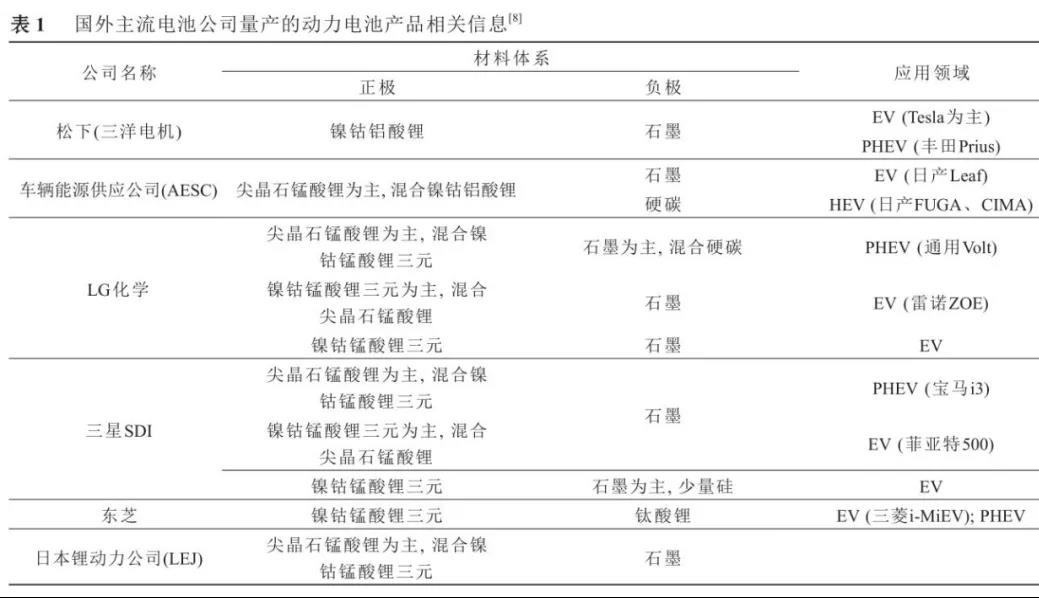
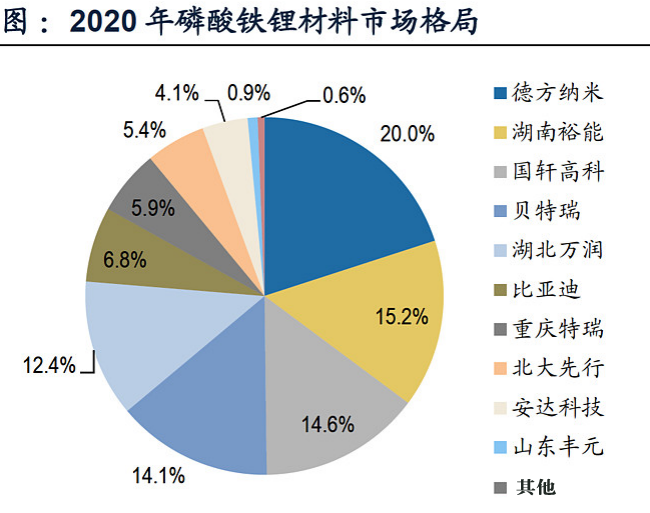
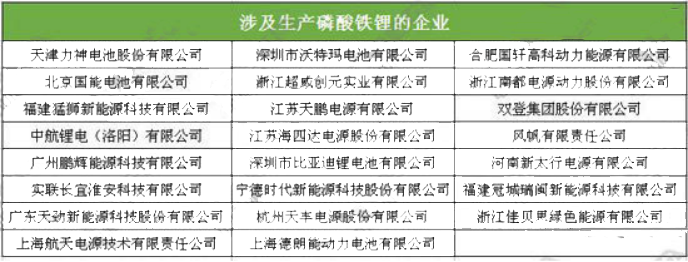
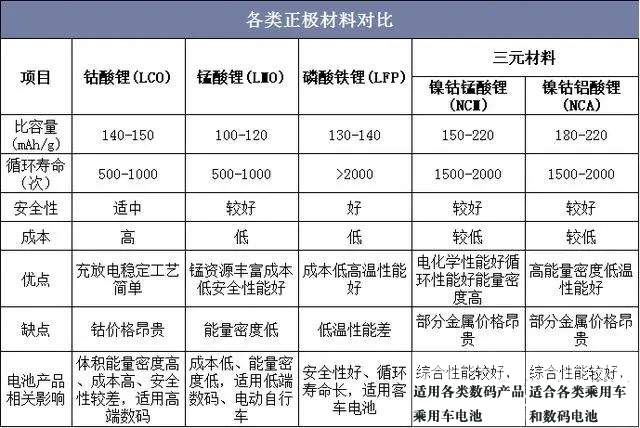
5、磷酸铁锂生产工艺研究方向与选择:LiFePO4生产工艺目前主要有高温固相反应法、碳热还原法、水热合成法、溶胶凝胶法、液相共沉淀法、微波合成法等;这些工艺都有各自的优缺点,但目前通过改良工艺后,应用比较广泛的还是前3种,国外多家公司采用固相法,美国的Valence公司采用碳热还原法,LG化学利用连续水热合成法;目前国内外已经能实现磷酸铁锂电池量产的合成方法主要是高温固相法,高温固相法又分传统的(以天津斯特兰、湖南瑞翔、北大先行等为代表,以草酸亚铁做为铁源)和改进的(以美国Valence、苏州恒正为代表,以三价铁物质做为铁源,该法也称碳热还原法)两种。对碳热还原法来讲,选取的铁源主要有两种,一种是Valence的氧化铁红路线,还有一种是清华大学(已成立北京锂先锋科技)以及武汉大学(已转让浙江振华新能源)的技术,选用磷酸铁做为铁源,该法制程工艺较为简单,其最大优点是避开了其它合成方法中使用磷酸二氢铵为原料,产生大量氨气污染环境的问题,但对磷酸铁原料要求较高。目前清华大学的一个研究小组通过控制沉淀条件合成了一种粒度可控,碳掺杂的磷酸铁前驱体,但该法合成难度较高,在工业放大过程中面临一些问题。
1)目前有些厂家选用磷酸二氢锂做为生产磷酸铁锂的原材料,同样可以避免反应过程的污染问题,这个在氧化铁红路线上有所体现;这条路线和磷酸铁加碳酸锂的路线均不产生污染。
2)在材料制备过程中,导电碳包覆是LiFePO4制备过程中的一项关键技术。A123通过在箔体表面预先涂敷一层高品质导电碳层,有效的降低了电池的内阻,提升了磷酸铁锂电池的大倍率放电能力。
3)LiFePO4正极材料具有循环性能好、比容量高、安全性能好以及原料来源广、价格低廉的特点,是下一代动力锂离子电池的首选材料。随着锂离子电池越来越广泛的应用,LiFePO4正极材料日益受到人们的关注国内外关于其结构性能以及制备改性的研究已经取得了巨大的发展,但对其制备改性的研究仍将是以后研究的重点。
4)LiFePO4材料的合成难度很大,目前所应用的主要是固相法生产,生产周期长、能耗高,污染严重,产品批次稳定性差,而且专利技术掌握在外国手中;面临知识产权的问题。为了实现LiFePO4材料生产的高效、节能,且稳定大规模的生产。国内必须研发出一种全新的技术工艺路线来实现磷酸铁锂材料的产业化。
5)近几年来我国开展锂离子电池正极材料研究开发的单位主要有:天津电子18所、北京有色金属研究总院、四川省有色冶金研究院、中科院化学所及物理所、中国兵器工业第二一三研究所、中南大学、厦门大学、中科院盐湖所、北京科技大学、清华大学、武汉大学、浙江大学、江西理工大学、东北师范大学等等单位。
6)国内用溶胶-凝胶法制备出前躯体,然后采取微波烧结的工艺路线,是我国现有的动力电池技术的一次大突破,技术达到国内先进水平,国际上亦未见报道;具有自主的知识产权,可以有效地提高产品的各项性能指标,保证产品的质量稳定,环保节能,大幅度地降低了生产成本。与目前国内外采用的工艺(固相法)相比,可节能40%以上,生产周期缩短50%以上。成本降低70%以上。

高镍三元正极材料生产工艺
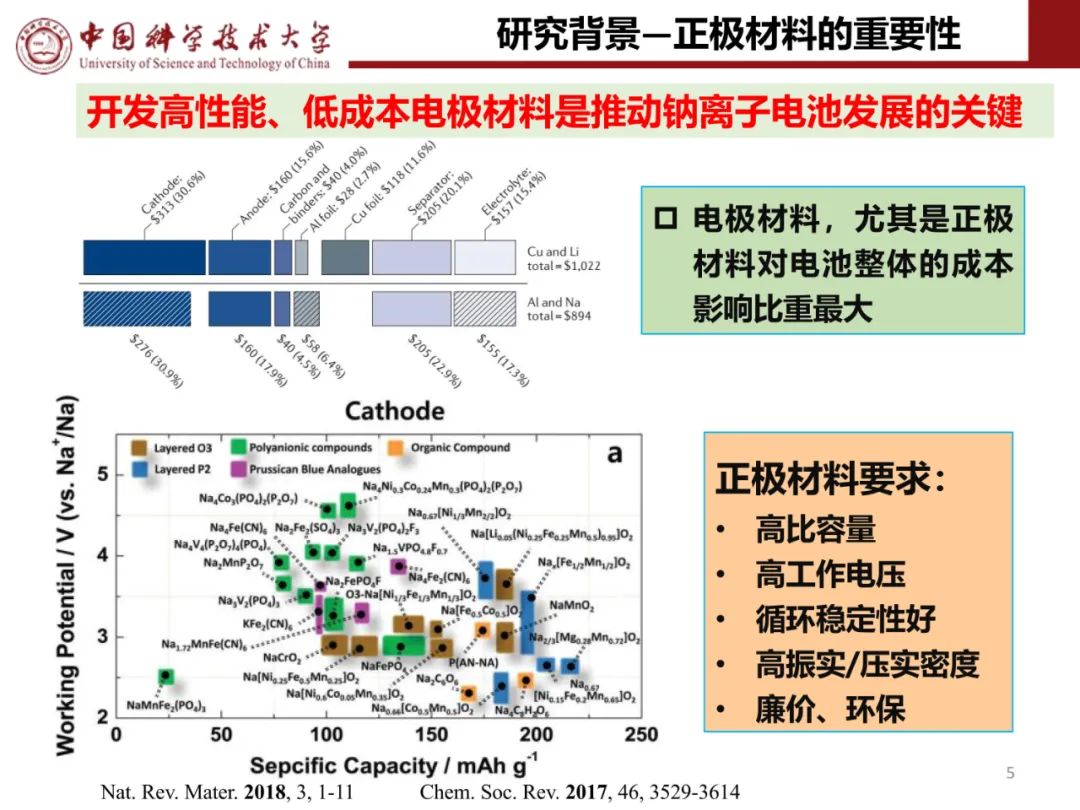
锂电池正极材料是锂离子电池构成材料的一部分,锂离子电池的主要构成材料包括电解液、隔离材料、正负极材料等。正极材料占有较大比例,因为正极材料的性能直接影响着锂离子电池的性能,其成本也直接决定电池成本高低。

高镍三元正极材料的主要工艺流程包括如下:1、锂化混合,2、装钵、3、煅烧、4、粉碎、5、分级、6、除杂、包装等。
1、锂化及混合:三元正极材料主要原料包括前驱体及锂源。混合方法一般分为湿法混合与干法混合。工业上多采用干法混合。高镍三元正极材料所使用的锂源为电池级单水氢氧化锂。在混料过程中,高镍三元材料要求混合更加均匀,对于湿度的控制也更为严格。
2、装钵:高镍三元正极材料所需锂源在前驱体中分散极好,为了防止材料在烧结过程中的自发团聚,每钵装料不能过多。
3、煅烧:煅烧工艺是高镍三元材料中最为核心的工艺,通常需要经过预烧和多次烧结。一般来说,镍含量越高,烧结温度越低。高镍三元材料实际烧结温度在750~800℃之间,烧结过程中需要使用纯氧辅助,富氧条件下反复多次烧结,煅烧时间普遍较长,一般为20~25h,因此其成本远高于普通三元材料。
4、粉碎与分级:高镍三元材料在煅烧后需要将颗粒破碎至微米级,通常需要经过三级破碎。目前工业常用的破碎体系为鄂式破碎-辊式破碎-气流破碎,获得颗粒粒度在1~20μm。经分级筛分,工业普遍选取D50=10μm级颗粒作为半成品颗粒。
5、除杂与包装:高镍三元材料颗粒分级后的半成品需要经过除杂,以除去工艺中引入的金属杂质,主要以铁为主,工业上通常使用磁选方式除铁。经除杂后的颗粒通过真空包装,形成正极材料颗粒成品。
层状氧化物锂电正极储钠性能



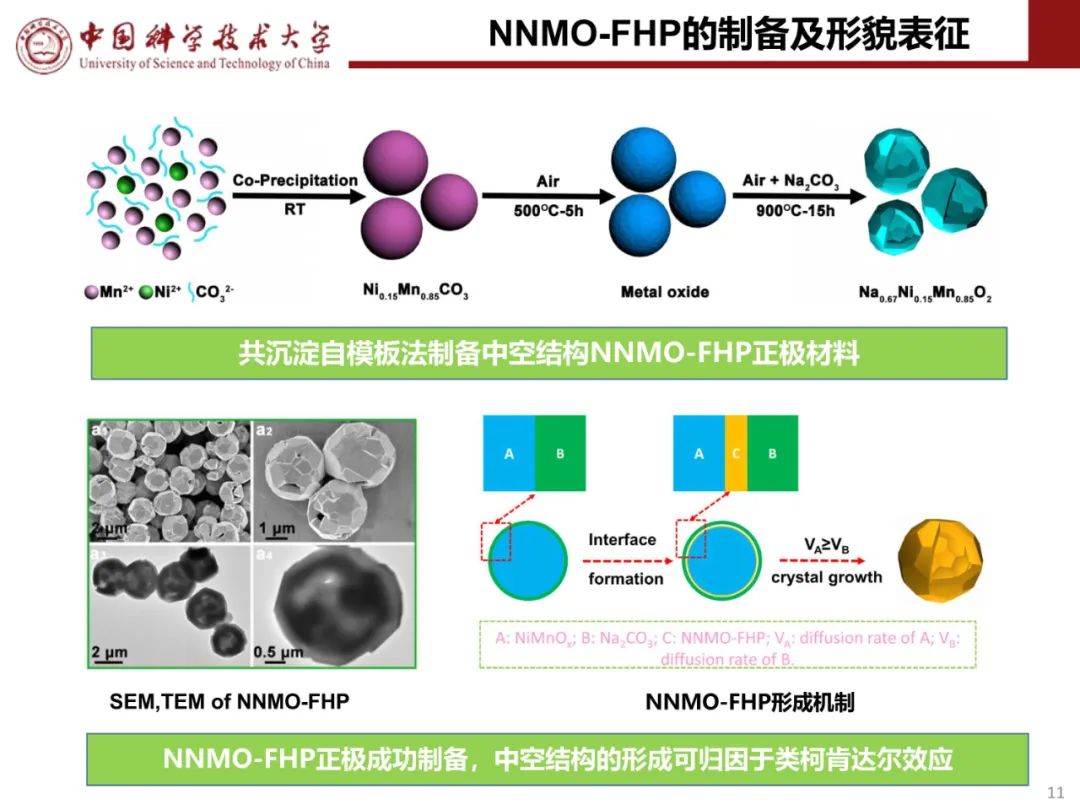
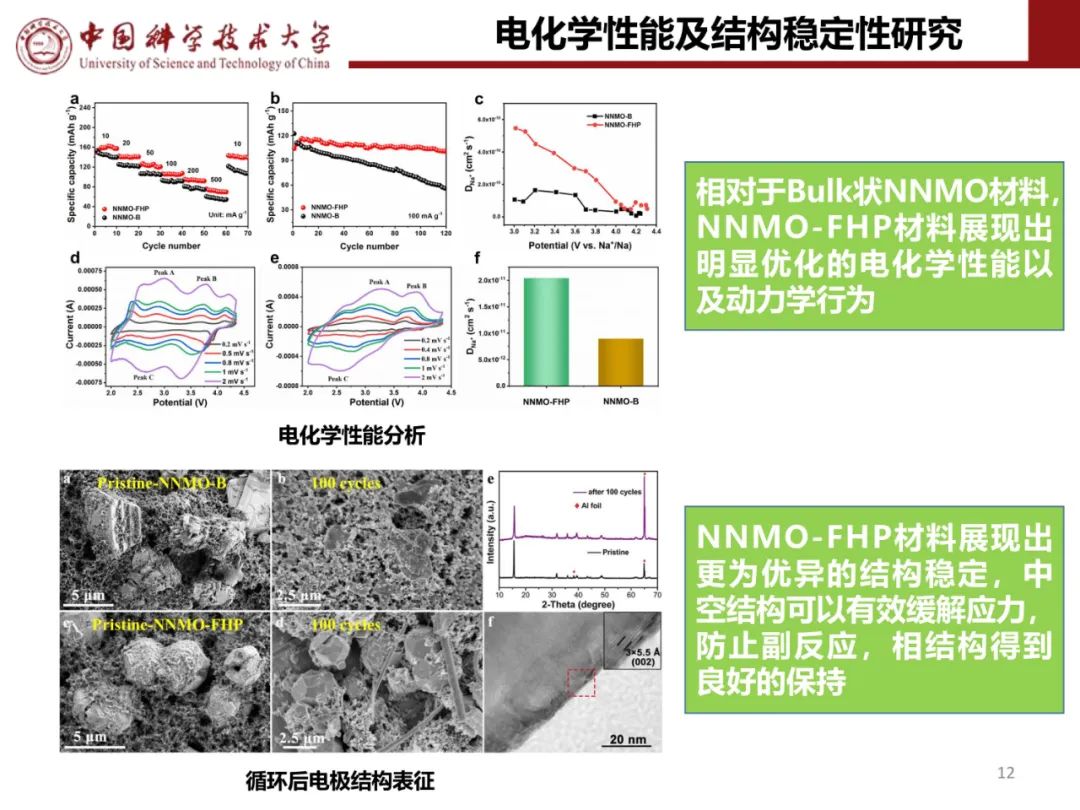


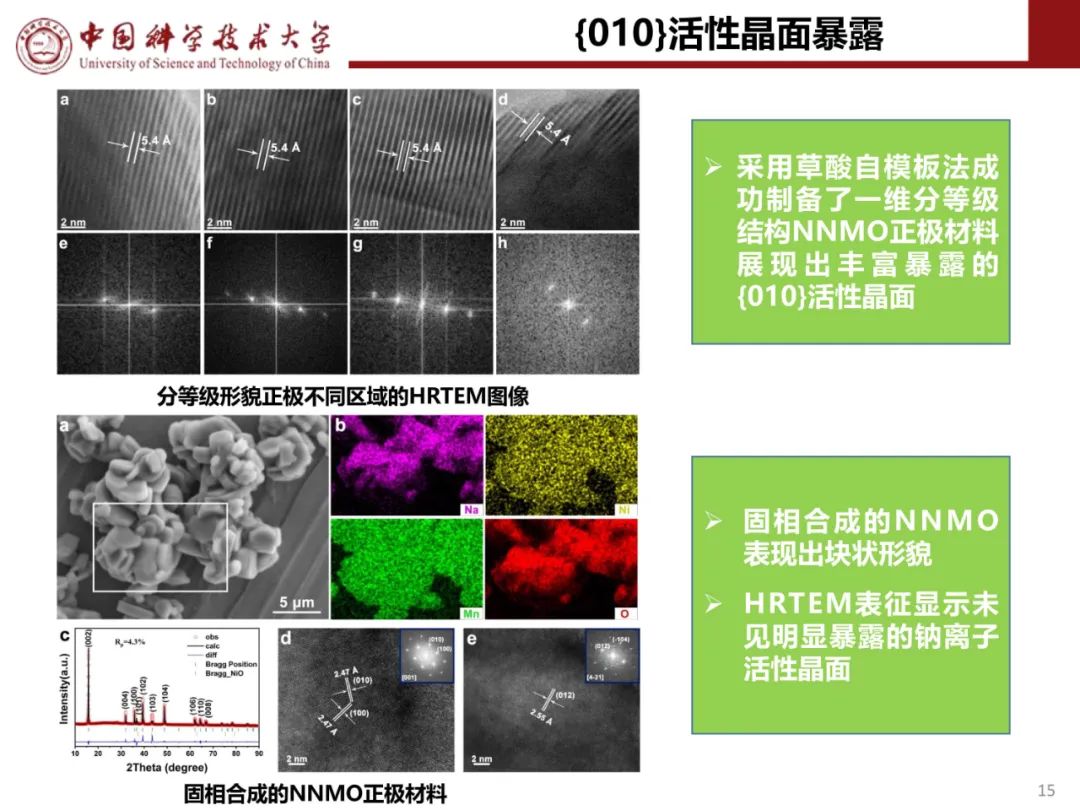
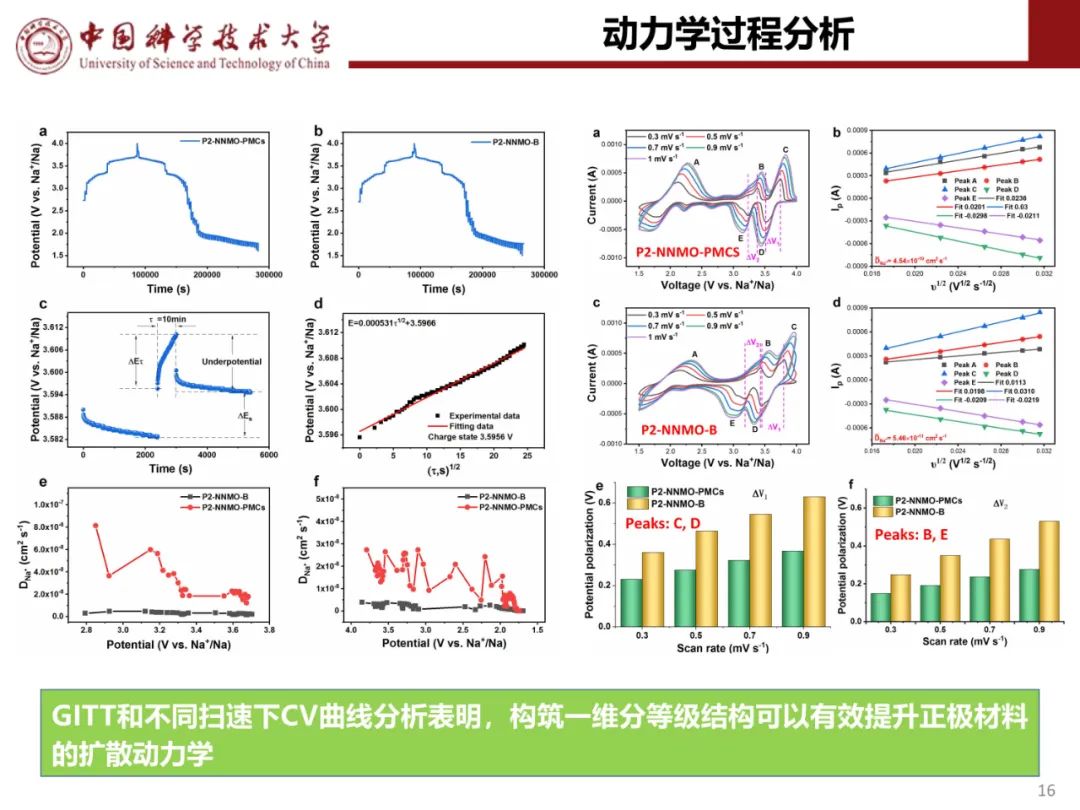
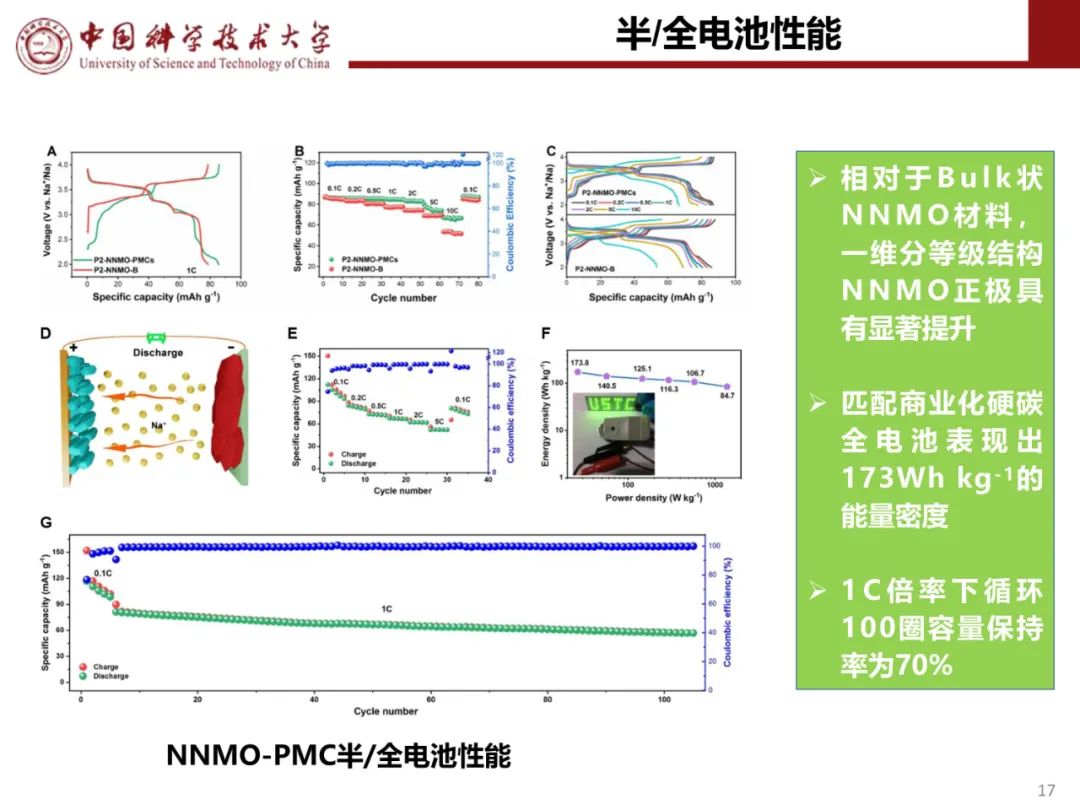
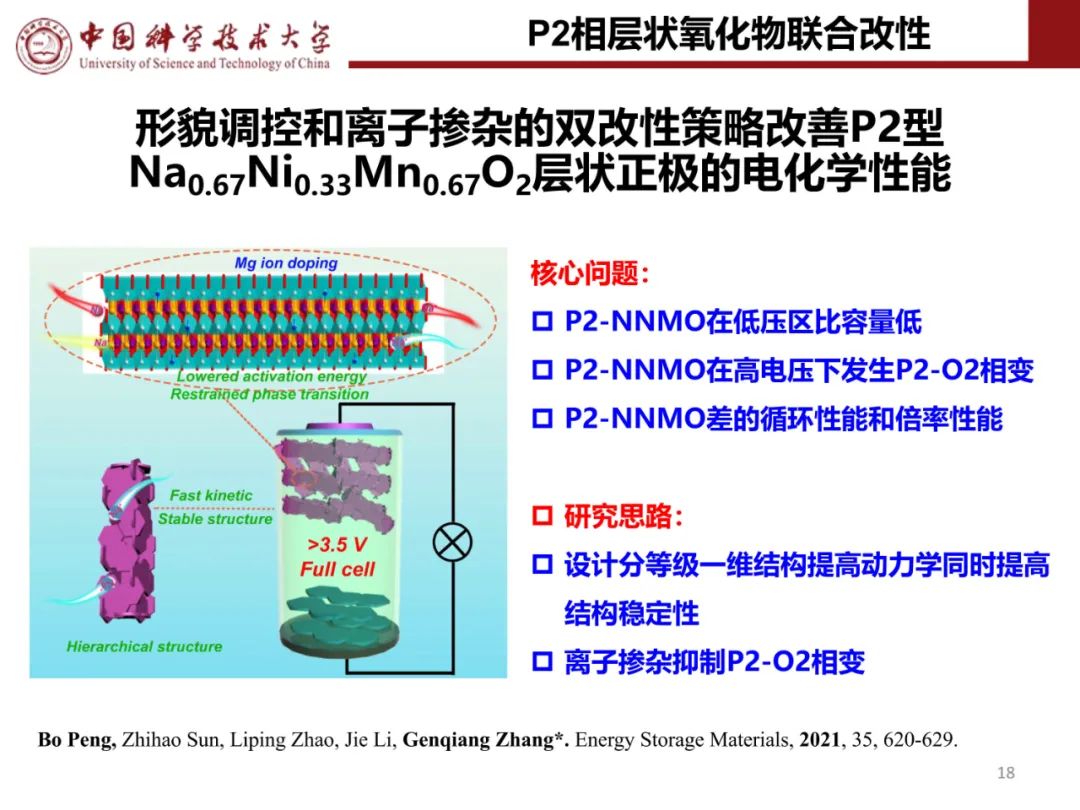
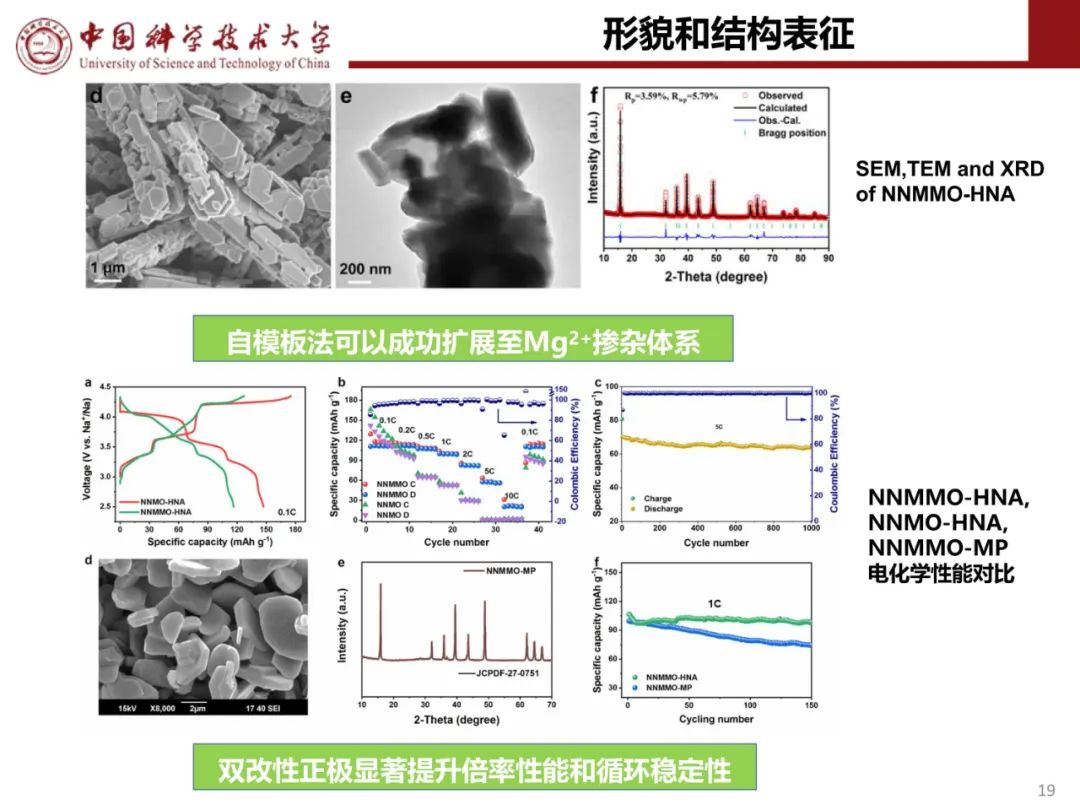
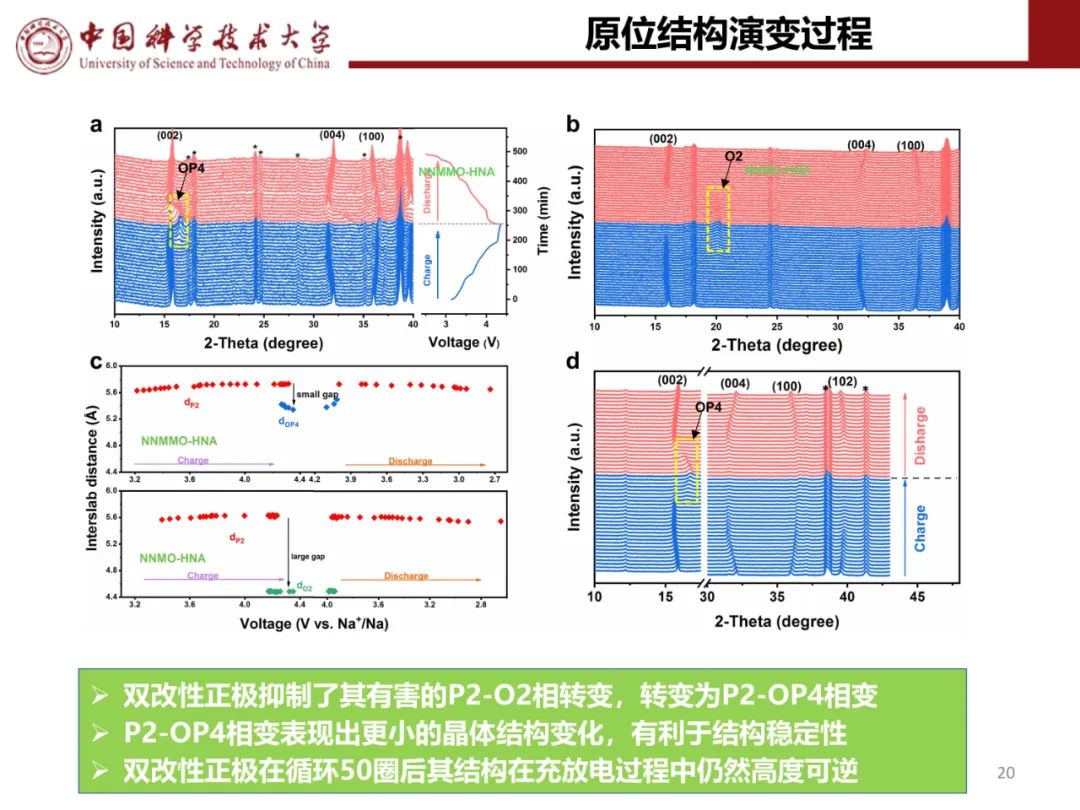
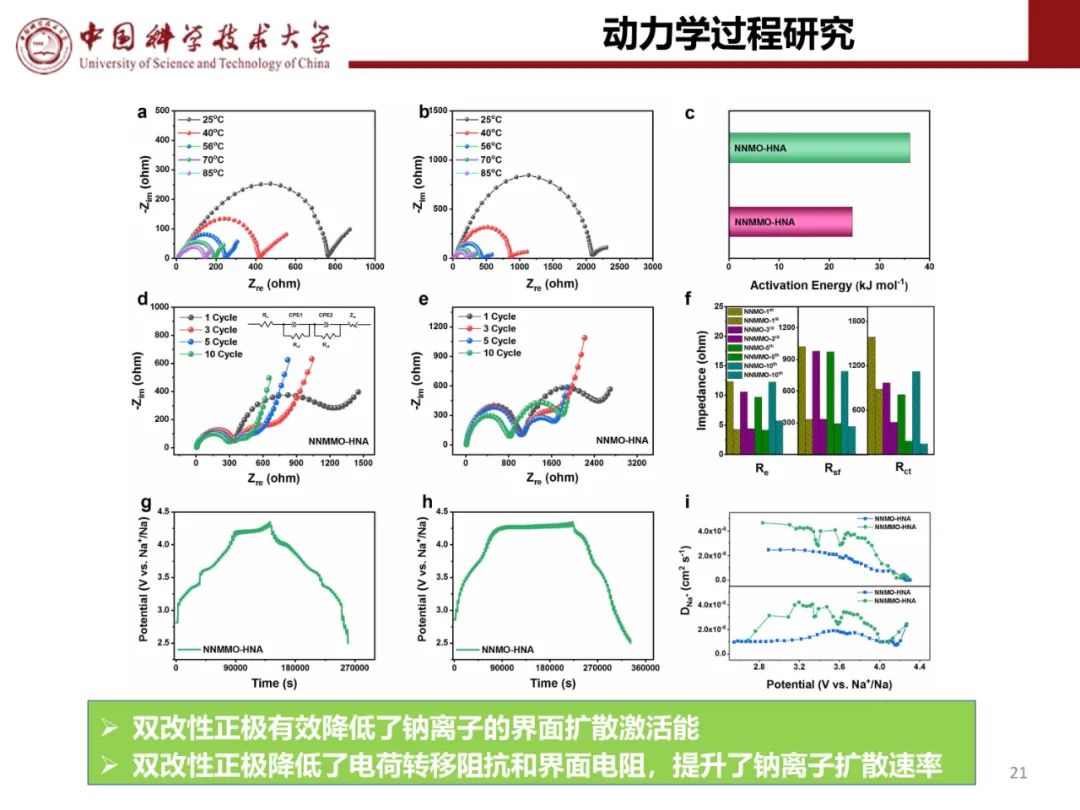
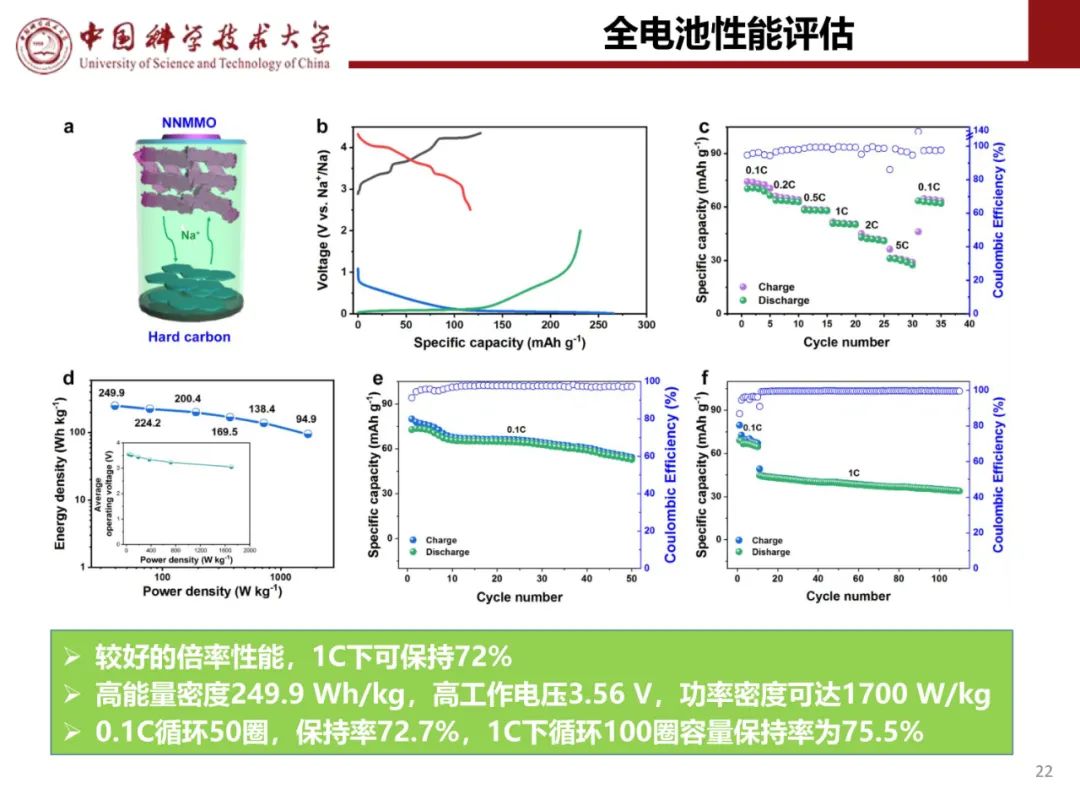
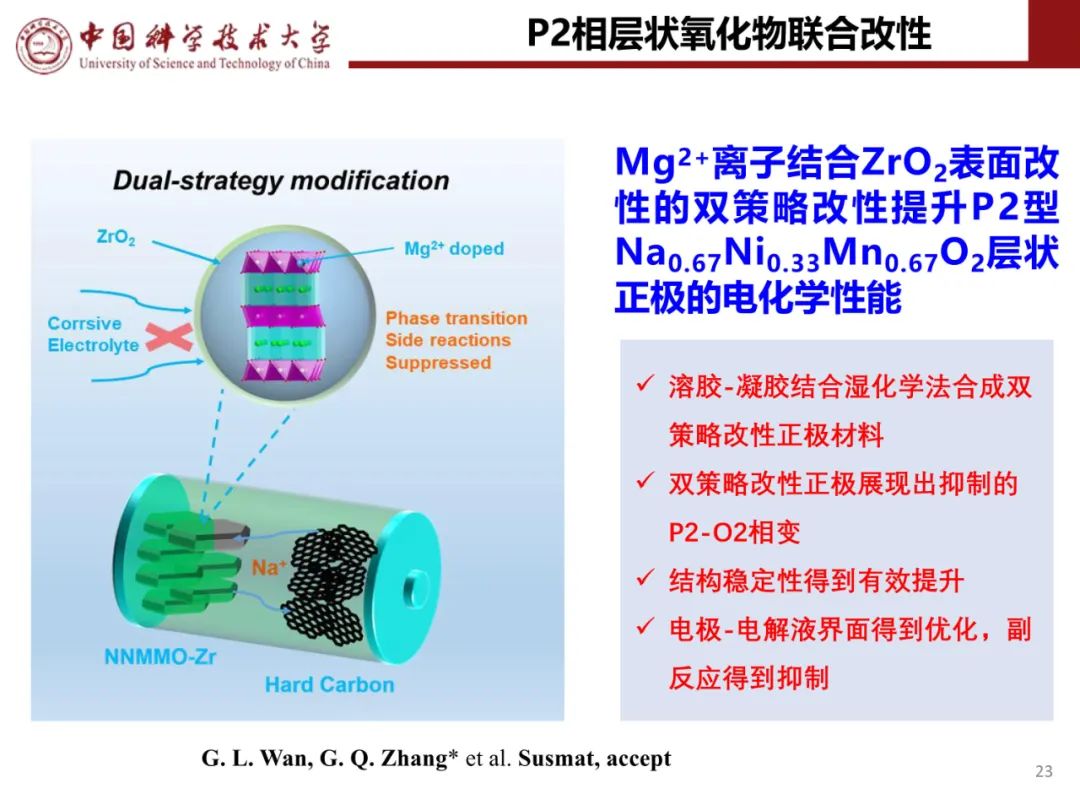
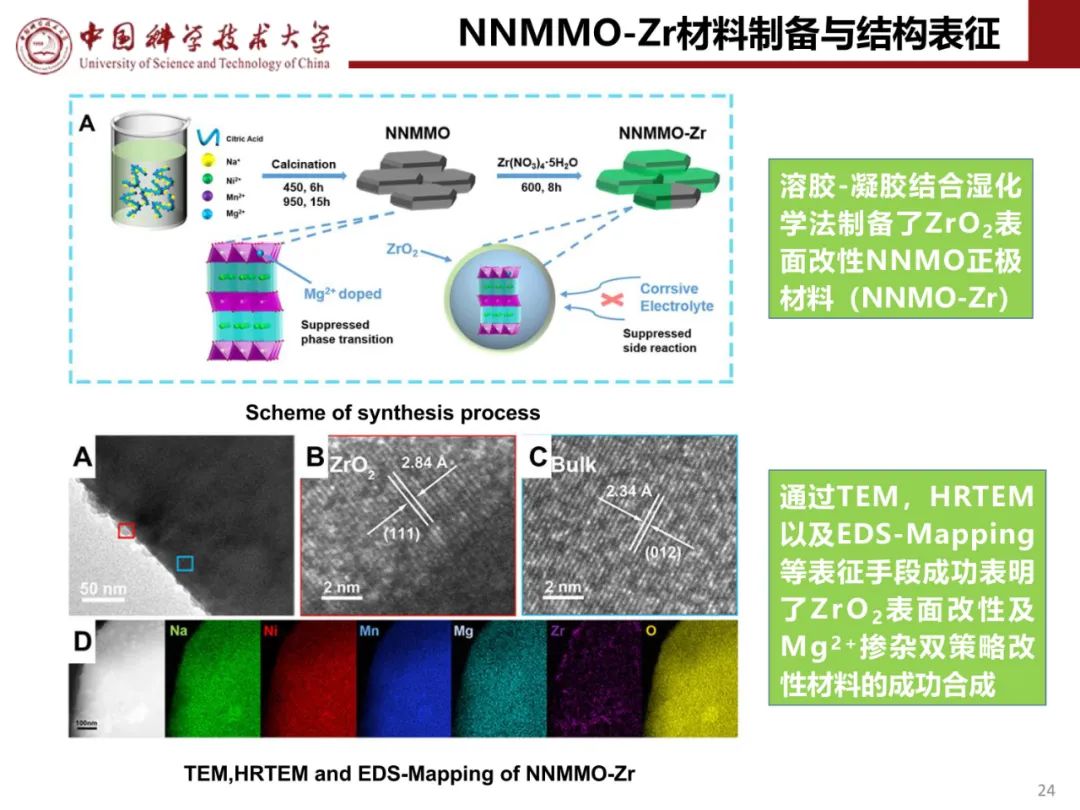
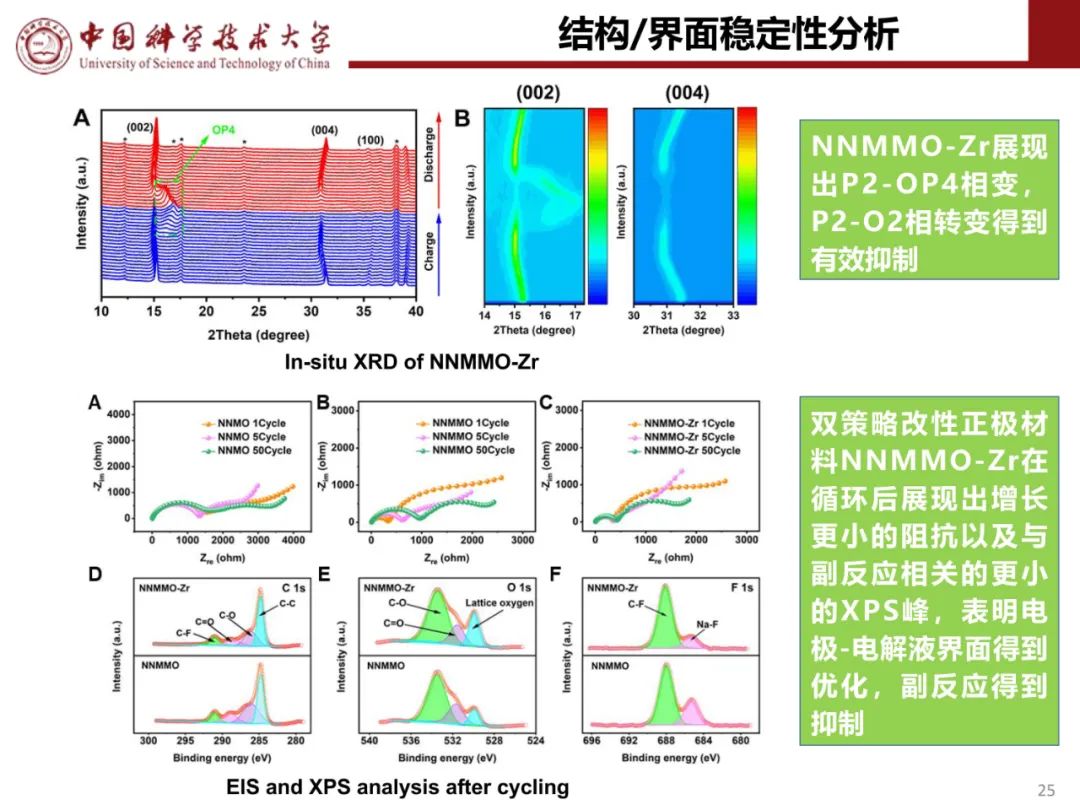
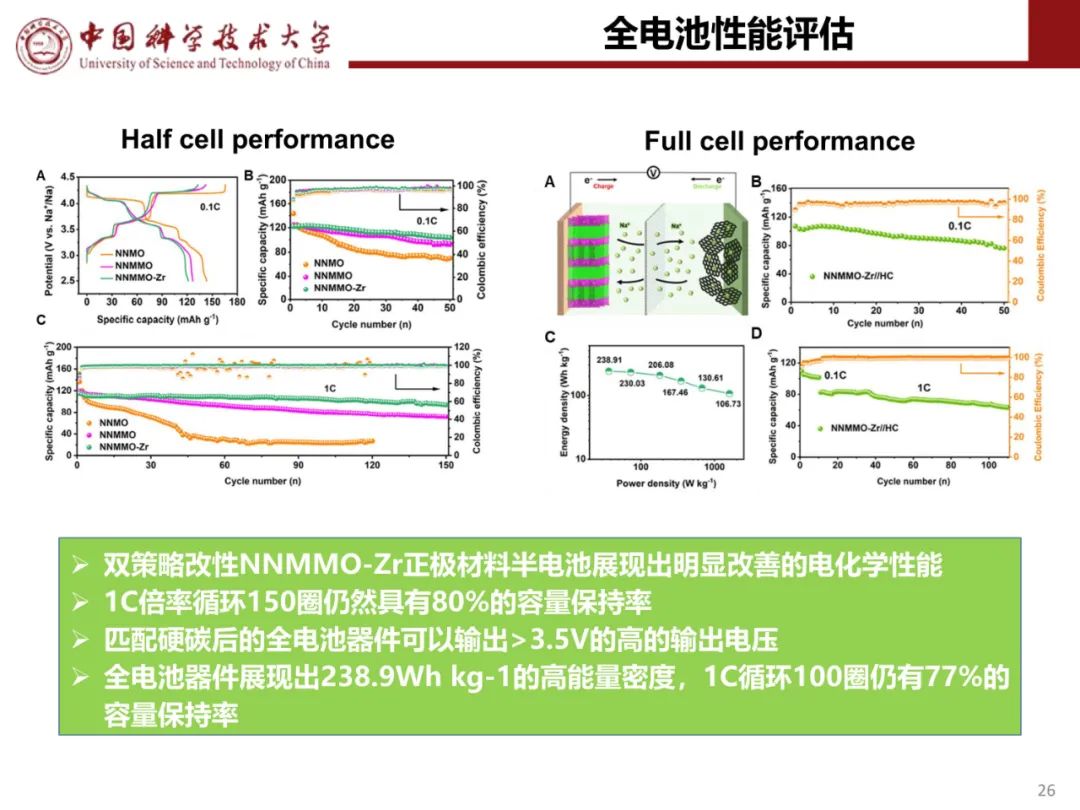

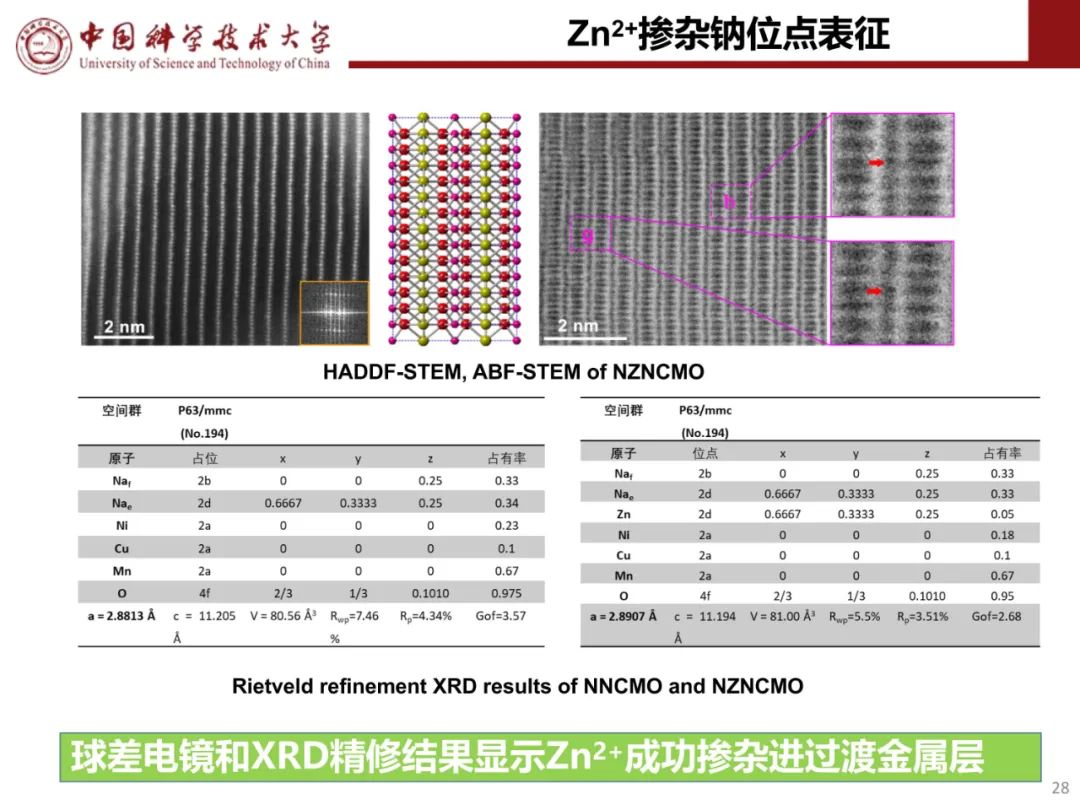
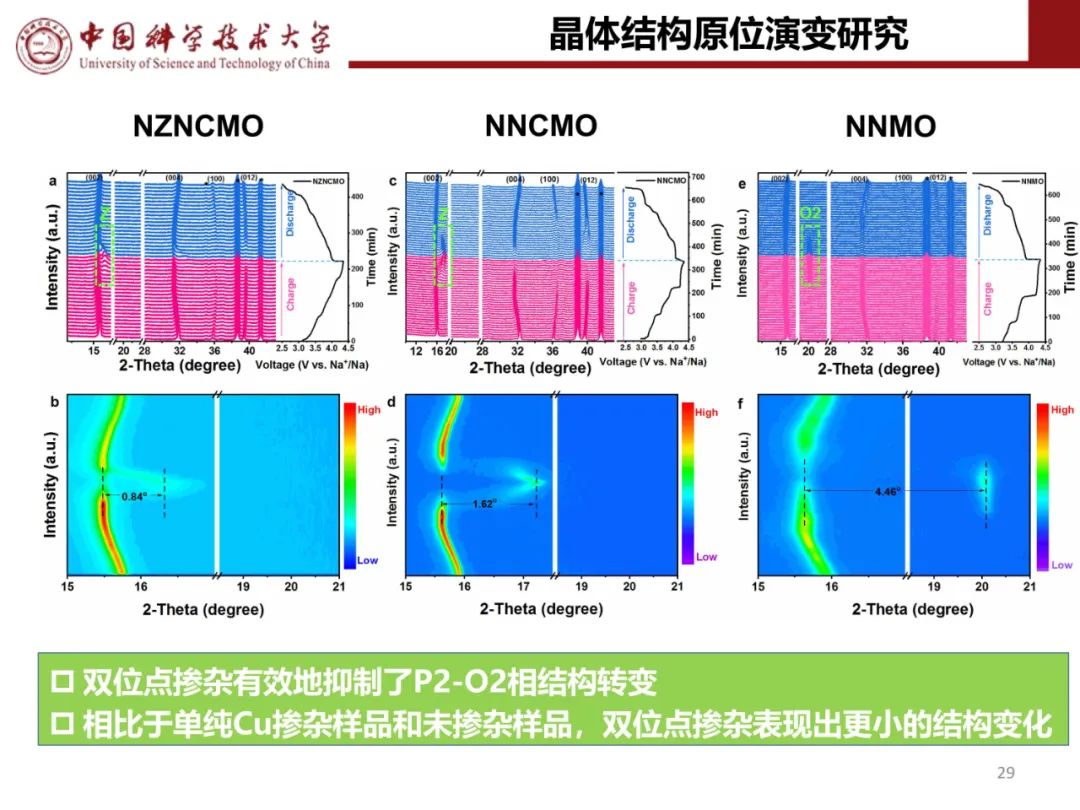
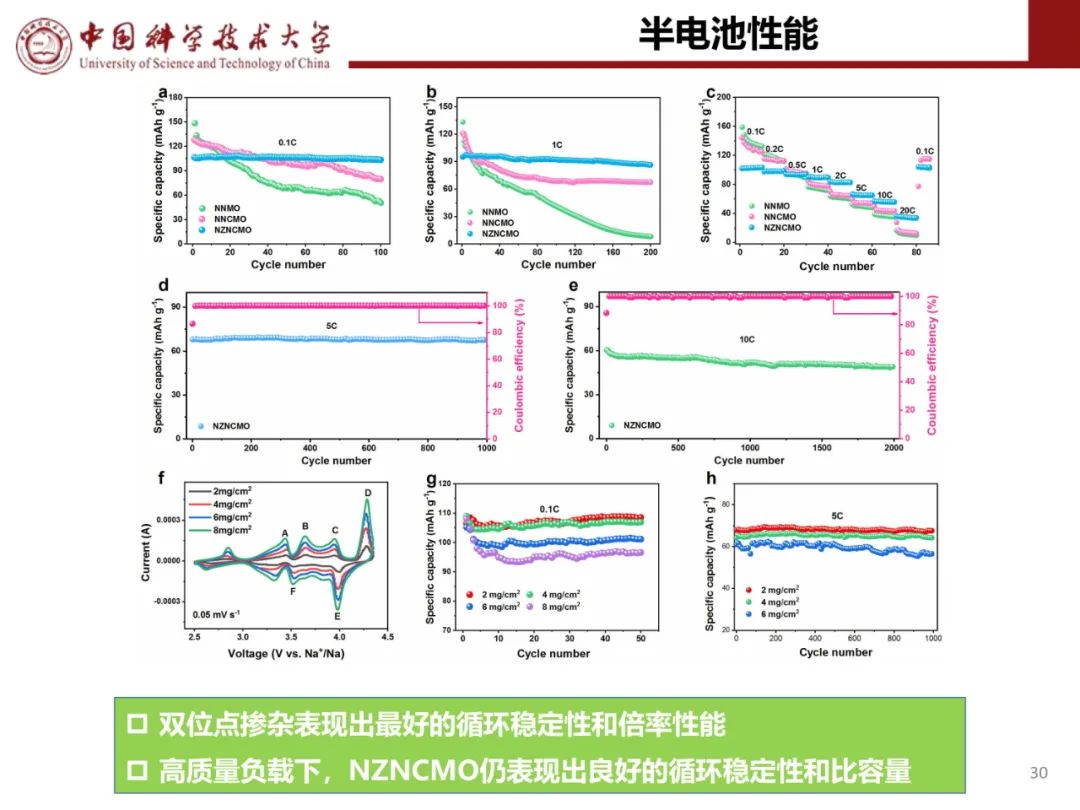
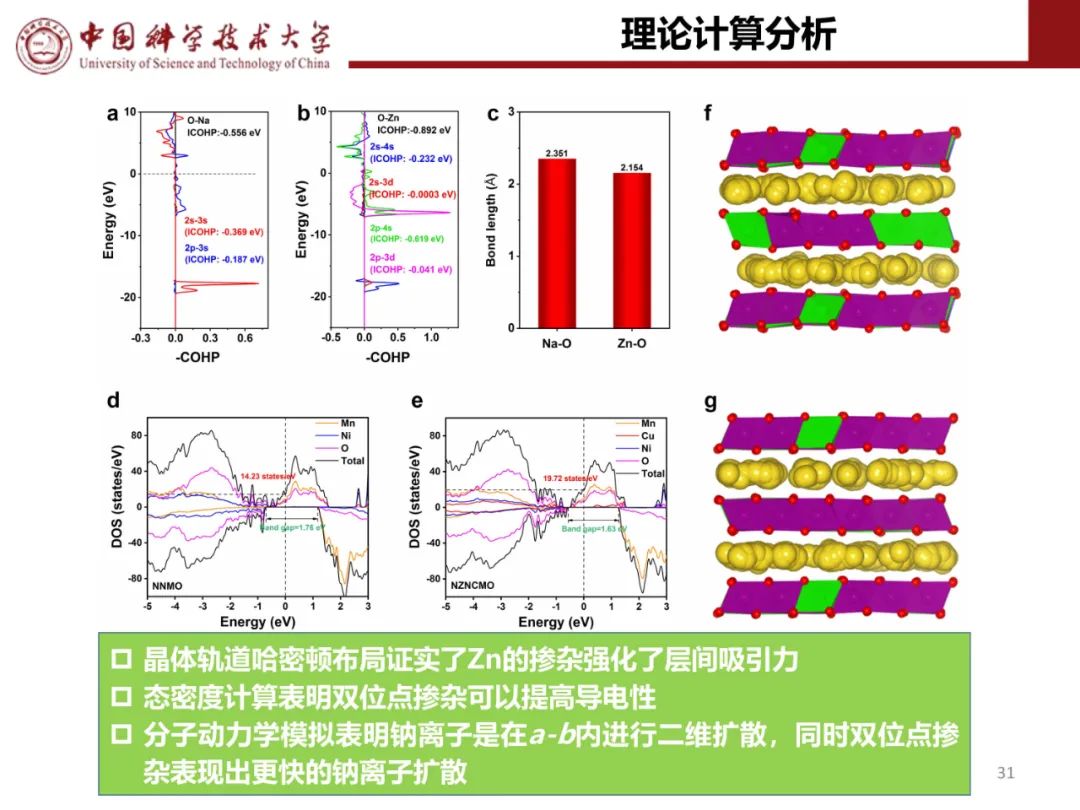
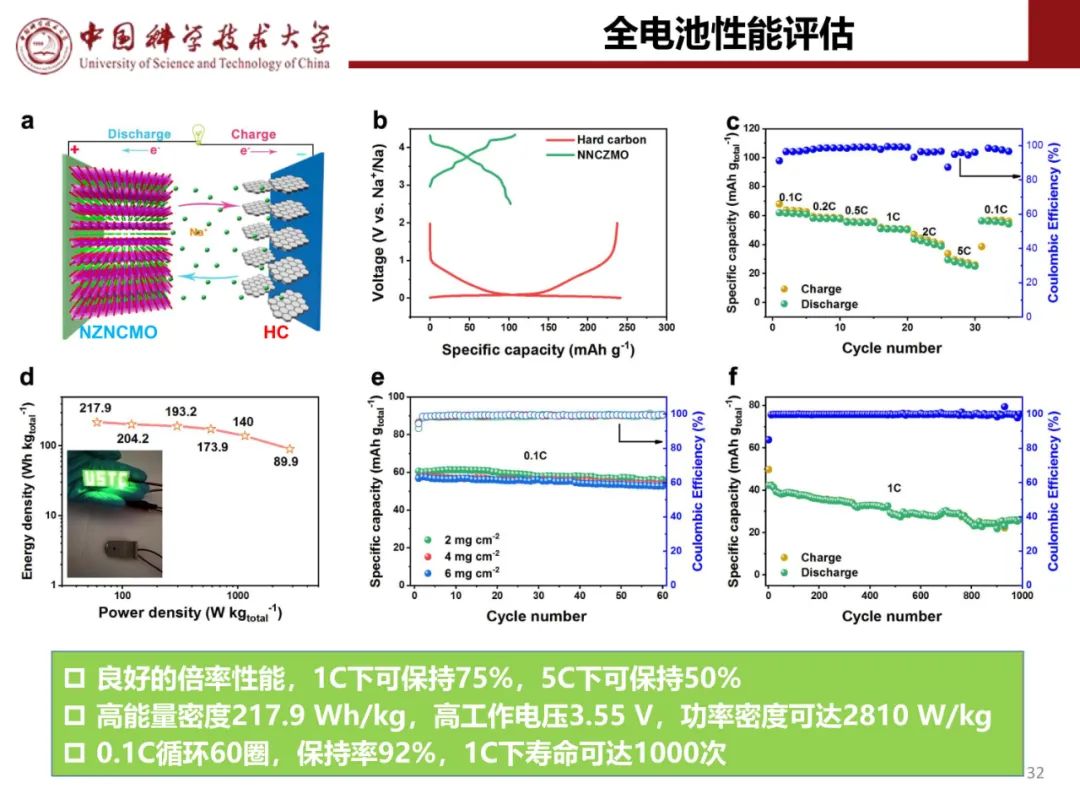
钠离子电池通用规范



























江苏翔鹰新能源科技有限公司成立于2016年6月,位于江苏溧阳市中关村科技产业园。公司目前注册资金为11250万元,项目总投资7亿。公司大股东为苏州江南嘉捷电梯有限公司。
溧阳市地处长三角中心“福”地,陆路与南京、杭州有高铁连接、与上海、苏州、无锡、常州、南京、杭州有高速公路连接。紧邻南京禄口国际机场航空港,与国际、国内大中城市有定期航班,交通非常便利。
公司坚持绿色能源理念发展新能源与新材料,以专业化人才加强科技创新,按照现代化企业制度规范化运作,不断开拓新能源材料的新领域,努力实现高科技、高品质新能源材料的产业化,为中国实现环保经济、蓝天白云常态化、健康发展贡献力量。
公司2016年12月建设完成的产能为3000吨/年,2017年6月扩产到10000吨/年,2018年6月已扩产到12000吨/年。公司开发和生产产品包括:锂电高镍三元,钠电层状氧化物正极材料。公司3000吨/年钠电层状氧化物正极材料于2022年9月1日正式投入生产,并于9月中旬批量供货。

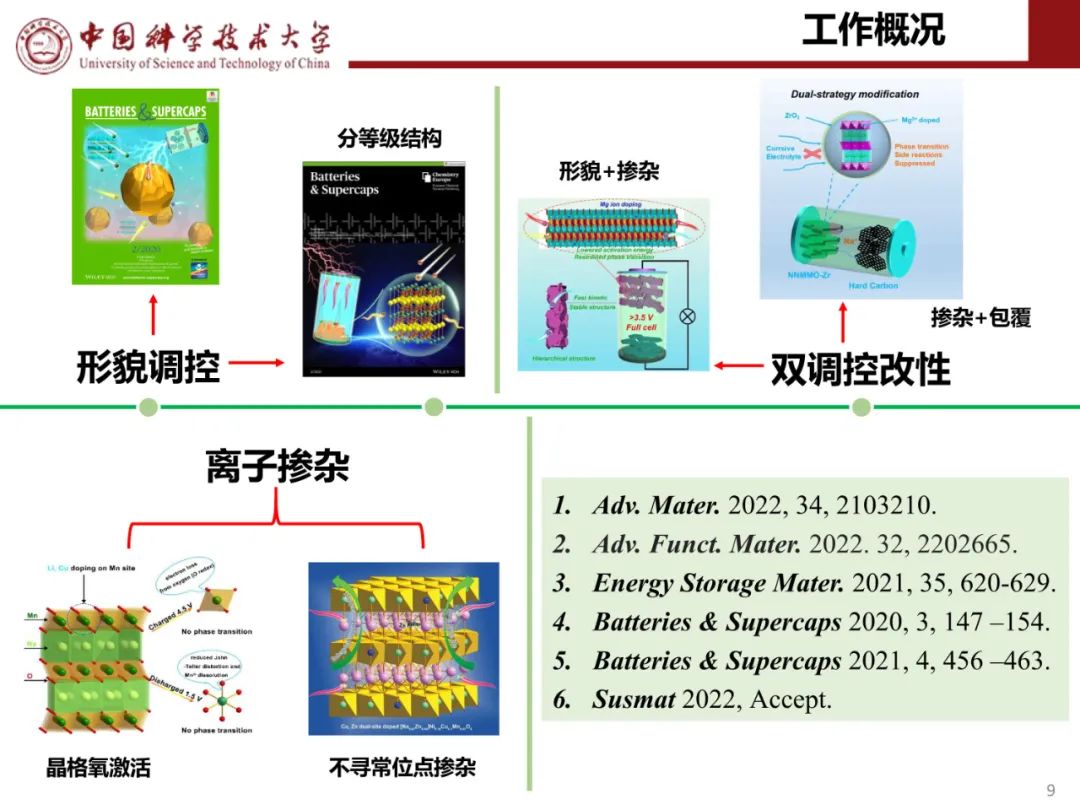
钠电正极:新型P2相铜锰材料在层状氧化物正极中激活阴离子氧化还原化学是设计高比能钠离子电池的典型方法。不幸的是,过量的氧氧化还原通常会引起不可逆的晶格氧损失和阳离子迁移,导致容量和电压快速衰减和反应动力学缓慢。
北京科技大学刘永畅教授,揭示了一种新型P2-Na0.8Cu0.22Li0.08Mn0.67O2正极中罕见的电子从氧转移到铜离子的还原偶联机制(RCM),以提高阴离子氧化还原反应可逆性和动力学。由此形成的强共价Cu-(O-O)键能有效抑制过量的氧氧化和不可逆阳离子迁移。因此,P2-Na0.8Cu0.22Li0.08Mn0.67O2正极提供了优异的倍率容量(在0.1C和100C时分别为134.1和63.2 mAh g-1)和出色的长循环稳定性(在10C下500次循环后容量保持率82%)。通过系统的原位/非原位表征和理论计算,充分了解了RCM的内在作用机制。该研究以题目为“Boosting the Reversibility and Kinetics of Anionic Redox Chemistry in Sodium-Ion Oxide Cathodes via Reductive Coupling Mechanism”的论文发表在国际顶级期刊《Journal of the American Chemical Society》上。

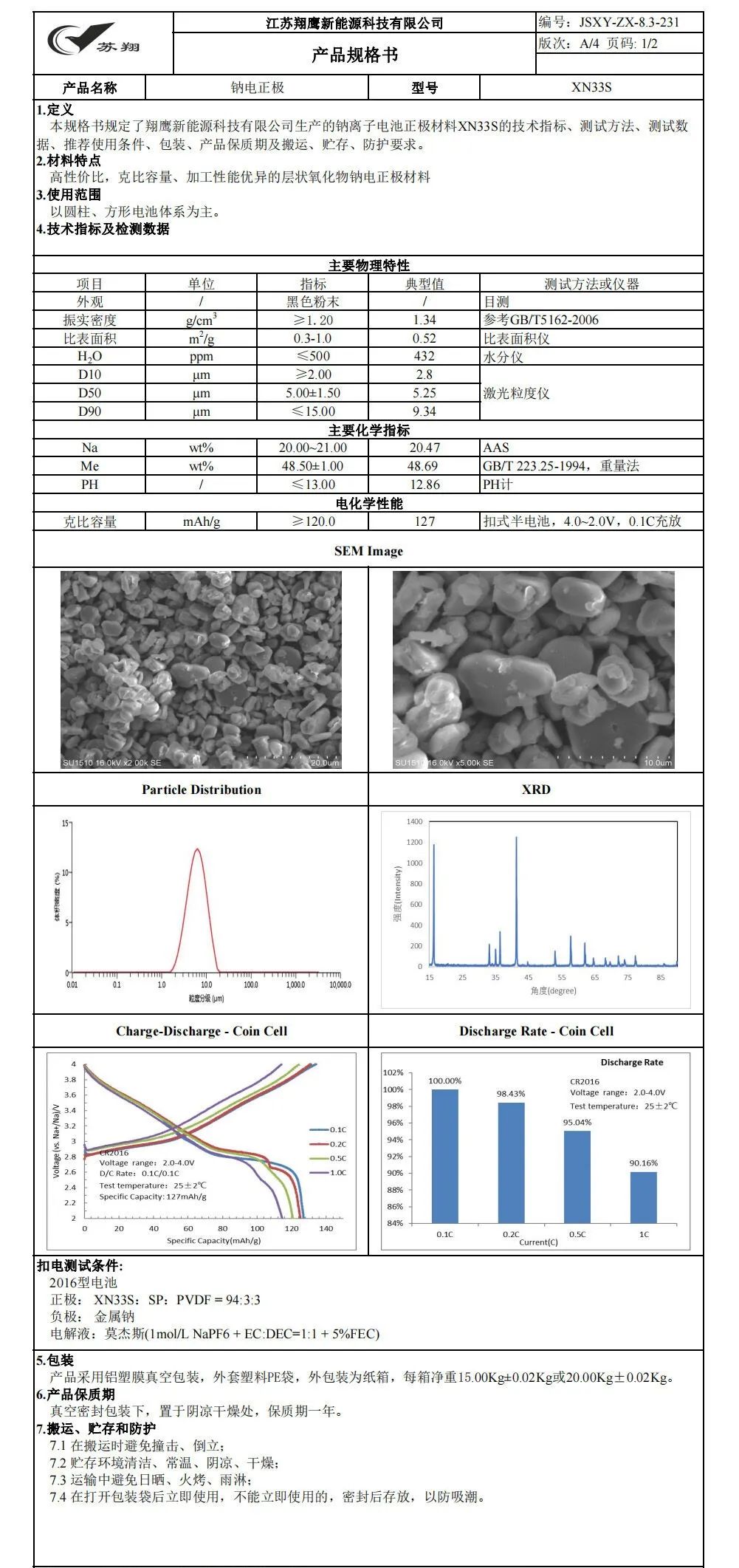
【图1】P2-Na0.8Cu0.22Li0.08Mn0.67O2的(a)XRD精修谱图和(b)7Li ss-NMR谱。(c)P2-NCLMO和P2-NZLMO晶体结构示意图。氧化物体系中Cu取代Zn的电荷密度差;(d)和(e)分别表示电荷的增加和减少。(f)P2-NCLMO和P2-NZLMO中O、Cu、Zn和Na离子的Bader电荷。(g)P2-NCLMO的SEM和TEM(插图)图像,(h)SAED图像,(i, j)HRTEM图像。(k)P2-NCLMO的Mn k边FT-EXAFS谱。(l)P2-NCLMO样品的SEM-EDS图谱。
采用溶胶-凝胶法制备了具有强Cu-O共价的P2型Na0.8Cu0.22Li0.08Mn0.67O2样品,并合成了具有离子Zn-O键的P2-Na0.8Zn0.22Li0.08Mn0.67O2材料(记为P2-NZLMO)进行比较。精修XRD图表明,P2-NCLMO材料在六方P2相(空间群:P63/mmc)中结晶,含有少量CuO(0.89%)杂质(图1a)。较小的R因子(Rwp=2.96%)表明实验曲线与计算曲线吻合较好。精修结果表明,由于离子半径相似,Li+主要被掺杂到TM位点。7Li固态核磁共振(ss-NMR)光谱也证实了这一点(图1b),其中大约84.5%的Li+位于TM位点(共振峰≈1640和≈1450 ppm),15.5%的Li+位于Na位点(共振峰≈750 ppm)。P2-NCLMO和P2-NZLMO的晶体结构如图1c所示,其中氧层沿c轴以ABBA顺序堆叠。Na离子占据两个不同的棱柱位,即Naf和Nae分别与TMO6八面体共享面和边。此外,P2-NCLMO的精修Na层间距(3.698 Å)大于P2-NZLMO的精修Na层间距(3.488 Å),有利于Na+的扩散动力学。这是因为Cu-O键的共价大于Zn-O键,会导致O离子周围的有效负电荷更低,从而降低Na与O离子之间的静电吸引力,导致Na-O键更长。电荷密度差分析表明,氧化物体系中以Zn取代Cu后,O离子和Zn离子的电荷密度分别增加(图1d)和减少(图1e),Bader电荷计算也得出了类似的结果(图1f)。扫描电镜(SEM)和透射电镜(TEM)图像显示,P2-NCLMO材料为片状颗粒,尺寸为1-5 μm,厚度为0.3-1 μm(图1g)。沿[001]晶带轴的选区电子衍射(SAED)图显示出明亮的衍射斑点,揭示了P2-NCLMO的单晶六方结构(图1h)。此外,高分辨率TEM(HRTEM)图像中具有良好分辨率的晶格条纹,其面间距分别为0.56和0.23 nm,对应于层状结构的(002)和(102)面(图1i,j)。值得注意的是,在(102)平面上可以直接观察到TM空位。Mn k边傅里叶变换扩展X射线吸收精细结构(FT-EXAFS)光谱如图1k所示。值得注意的是,第二配位壳中Mn-TM相互作用的配位数(CN)为5.6,低于标准六方配位值6.0,进一步证实了TM空位的存在。SEM能谱图(EDS)表明,Na、Cu、Mn和O元素在P2-NCLMO颗粒中均匀分布(图1l)。
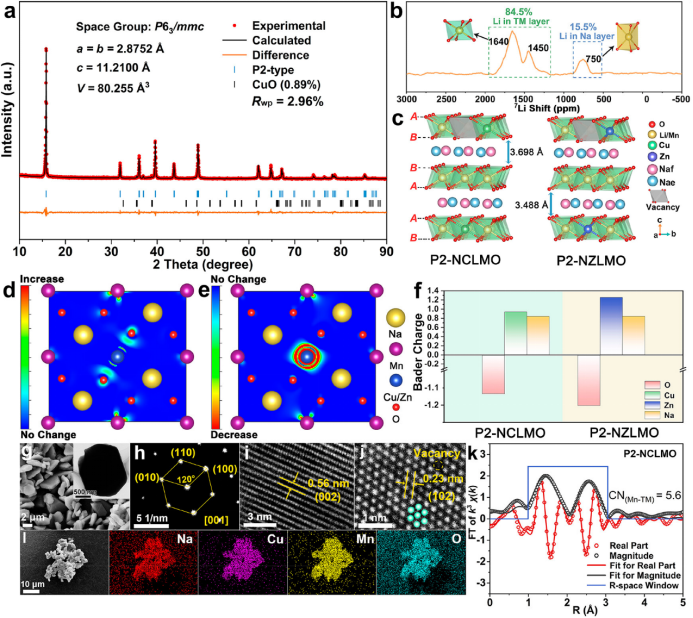
【图2】(a)0.2 mV s-1下的CV曲线和(b)0.2C时P2-NCLMO正极的恒流充放电曲线。(c)P2-NCLMO和P2-NZLMO在0.2C时的循环稳定性和(d)电压滞后。插图显示了两个样品经过50次循环后的HRTEM图像和相应的GPA图像。(e)P2-NCLMO和(f)P2-NZLMO从0.1C增加到1C时的放电曲线和相应的dQ/dV图。(g)P2-NCLMO在5C和10C下的倍率性能和(h)长循环稳定性。插图显示了本研究与报道的Cu–Mn基钠层状氧化物的比较。
图2a绘制了P2-NCLMO在0.1 mV s-1扫速下的循环伏安(CV)曲线,在2.18/2.00、2.44/2.35、2.83/2.77、4.03/3.91和4.40/4.27 V处发现了5对氧化还原峰。以4.03/3.91 V和4.40/4.27 V为中心的峰分别属于Cu2+/Cu3+和O2-/(O2)n−氧化还原反应,而3.4 V以下的峰与Mn3+/Mn4+氧化还原对有关。重叠良好的CV图表明高度可逆的氧化还原反应伴随着重复的Na+插入/脱出。P2-NCLMO电极在0.2C(1C=130 mA g-1,图2b)下的初始充电容量为90.4 mAh g-1,相当于约0.34个电子转移,超过了基于Cu2+氧化为Cu3+的理论容量(每公式单位0.22个电子转移)。由于四价锰不能被进一步氧化,并且所使用的电解质在4.5 V以上分解,因此额外的容量应归因于氧的氧化。此外,高压区重叠的充放电曲线反映了P2-NCLMO正极中稳定的阴离子氧化还原化学,这得益于Cu 3d态和O 2p态之间的强共价。对于P2-NZLMO电极,循环后CV曲线上的氧氧化峰非常尖锐但明显降低,与电压曲线上逐渐降低的充电平台相吻合,表现出更强但不可逆的氧氧化还原活性。这种现象可以解释为强离子Zn-O键在提供过多电子后会引起晶格氧损失。0.2C时P2-NCLMO和P2-NZLMO电极的循环性能和相应的平均放电电压变化如图2c所示。在50次循环后,P2-NCLMO正极的容量保持率为90.3%,平均放电电压更高且更稳定,而P2-NZLMO电极在50次循环后容量衰减迅速,容量保持率仅为57.2%,电压下降严重。循环后的HRTEM图像和相应的几何相分析(GPA)图(图2c插图)显示,P2-NCLMO的层状结构保持良好,内部应变小而均匀,而P2-NZLMO的内部应变巨大而密集,并伴有严重的晶格畸变和颗粒裂纹。以上结果证明,Cu-O的强共价可以抑制不可逆氧释放,保证结构的完整性。
此外,严重的电压滞后是氧氧化还原反应面临的另一个致命问题,它会降低电化学性能。与P2-NZLMO相比,P2-NCLMO正极在高压区域表现出更低的电压滞后(图2d),表明阴离子氧化还原动力学得到改善。倍率性能测试进一步证实了这一事实(图2e,f)。P2-NCLMO电极呈现出重叠的放电曲线,在高电压下相应的dQ/dV峰与倍率的增加吻合良好,而P2-NZLMO电极的放电容量衰减迅速,dQ/dV峰强随倍率的增加而明显降低。这是因为P2-NCLMO中的阴离子反应是由快速动力学阳离子反应(Cu2+/Cu3+)介导的。此外,Cu-O的强共价可以通过还原耦合机制有效地减缓TM的迁移,从而促进Na+的扩散动力学。因此,P2-NCLMO正极在0.1C和100C时的放电容量分别达到了134.1 mAh g-1和63.2 mAh g-1(图2g),打破了阴离子氧化还原正极通常具有较差倍率性能的传统观念。此外,P2-NCLMO的长循环性能如图2h所示,在5C下循环200次后容量保持率为90.1%,在10C下循环500次后容量保持率为82%。在最初的几个循环中,容量的增加与电极活化过程有关。图2g、h的插图显示,P2-NCLMO正极的倍率性能和循环稳定性在已报道的Cu–Mn基钠氧化物和其他涉及阴离子氧化还原化学的Na离子层状氧化物正极中脱颖而出。
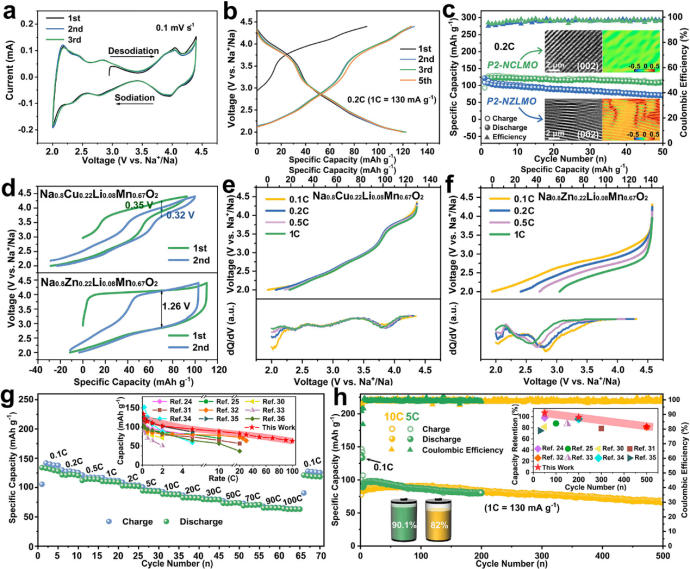
【图3】(a)0.2C第一次循环时P2-NCLMO电极的原位XRD谱图及相应的体积变化,(b)(002)峰等高线图。(c)P2-NCLMO正极和以前报道的SIB的P2型正极之间的最大单胞体积变化的比较。(d)Cu 2p XPS谱,(e)Cu L边EELS谱,(f)Cu K边XANES谱,(g)P2-NCLMO相应的FT-EXAFS谱。不同电压状态下(h)Cu和(i)Mn K边WT-EXAFS谱线图(j)不同电化学状态下P2-NCLMO的O 1s XPS光谱。(k)P2-NCLMO在原始状态、充电至4.15 V和充电至4.4 V时的EPR谱。(l)Cu 3d和O 2p轨道电子构型在初始脱钠过程中的演化示意图。
在2.0-4.4 V的电压范围内进行原位XRD测量,研究了P2-NCLMO的结构演变(图3a,b)。在充电过程中,(00l)峰逐渐向低角度区域移动,说明Na+脱出后相邻氧层间静电斥力增大,导致层间距扩大。同时,(101)峰不断向更高的角度移动,表明由于TM离子氧化导致ab平面收缩。在接下来的放电过程中,所有衍射峰都经历了可逆的移动,(001)和(101)峰比原始状态分别向更高和更低的角度移动,这是由于在初始循环中插入的Na+比脱出的Na+多。在整个充放电过程中,没有出现额外峰或原始峰消失/分裂,表明P2-NCLMO电极的固溶反应非常稳定。这是因为低价Li+掺杂确保了Na层中有足够的钠来稳定在深度充电状态下的P2结构,并有效地减轻了Na+/空位的有序化。P2-NCLMO的最大单胞体积变化仅为1.26%,远小于之前报道的P2型钠离子氧化物正极(图3c),说明P2-NCLMO具有出色的循环稳定性。
通过XAS、XPS、EELS和EPR分析系统地揭示了P2-NCLMO正极的电荷补偿机制。图3d为不同电化学状态下的Cu 2p XPS光谱。原始电极的Cu 2p1/2(952.8 eV)和Cu 2p3/2(933.1 eV)峰表明铜为二价态。当充电至4.15 V时,Cu 2p1/2和Cu 2p3/2峰分别跃迁到较高的结合能954.2和934.1 eV,表明Cu2+完全氧化为Cu3+。当完全充电至4.4 V时,Cu2+物种重新出现,表明Cu离子部分还原。这种反直觉的TM在充电至高压时的还原行为为还原耦合机理提供了证据。当放电到4.0V时,随着电子返回到O离子,Cu 2p峰异常地移动到更高的能量,然后在放电到3.4V时恢复到原始状态,这表明Cu离子具有高度可逆的还原耦合机制。非原位Mn 2p XPS光谱显示,原始四价Mn离子主要受低价Li+和Cu2+掺杂控制,在初始充电过程中电化学活性不强,但在低压区有助于电荷补偿。在循环过程中放电至2.0 V时,Mn的平均价态较高,说明P2-NCLMO存在较弱的Jahn-Teller效应。此外,还进行了非原位EELS测试,以监测Cu和Mn离子在充放电过程中的价态变化,结果与非原位XPS结果吻合良好(图3e)。进一步采用非原位XAS探测P2-NCLMO的电子结构变化。图3f、g显示了Cu k边的归一化x射线吸收近边结构(XANES)和EXAFS光谱。当充电至4.15 V时,Cu k边XANES谱明显向更高的能量移动,第一Cu-O配位层明显缩小,表明Cu2+发生氧化。当充满电至4.4 V时,随着Cu-O距离的减小,Cu k边谱向更低的能量移动。这是因为在深度脱钠状态下,通过电子从氧转移到铜离子,形成了高共价的Cu-(O-O)键。当放电至4.0 V时,由于电子返回氧,这种Cu-(O-O)键消失,导致Cu离子部分氧化,Cu-O距离增大。第二Cu-TM配位壳由于其共边八面体结构而发生了类似的变化。还记录了Cu和Mn k边的小波变换(WT)EXAFS光谱(图3h,i),直观地显示了在充放电过程中键长变化,其中1.5和2.5 Å附近的两个散射峰分别对应于TM-O和TM-TM键。
为了研究氧氧化还原在电荷补偿过程中的贡献,收集了非原位O1s XPS光谱(图3j)。在原始样品中观察到以529.5 eV(晶格氧)、531.3 eV和532.8 eV(表面氧)为中心的三个拟合峰。当充满电至4.4 V时,在530.5 eV处出现一个新的(O2)n−峰,表明氧被氧化。该峰在放电过程中消失,即使在10次循环后,也可以在充满电状态下再次检测到。精细扫描非原位EPR光谱进一步证实了Cu离子的氧氧化还原反应性和还原耦合机理。如图3k所示,原始样品的EPR谱呈以g=2.079为中心的各向异性线形,其中Cu2+(3d9)的电子自旋S=1/2与核自旋I=3/2耦合产生了4条超细线。当充电至4.15 V时,由于抗磁性Cu3+(3d8)的形成,无法检测到EPR信号。在完全充电至4.4 V后,Cu2+共振再次出现,并且可以清楚地观察到新的(O2)n-信号(g=2.008),与非原位XPS, EELS和XANES结果相吻合。图31描述了Cu - NCLMO正极中电荷从O向Cu转移形成Cu-(O-O)构型的还原耦合机制。
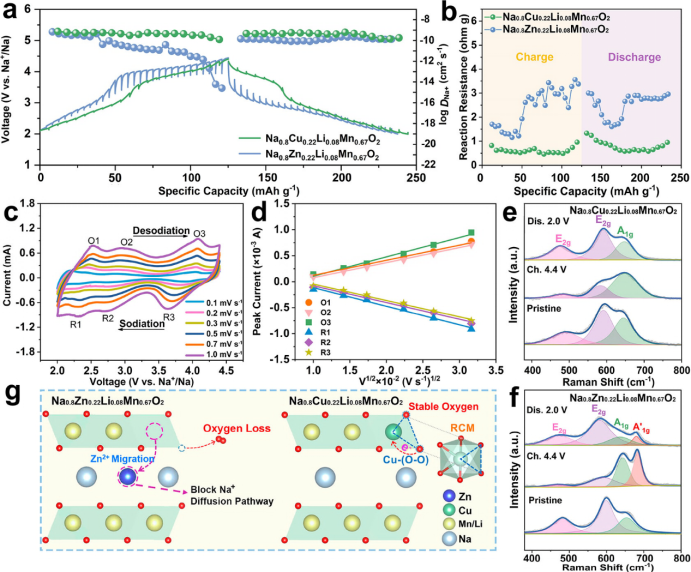
【图4】(a)0.2C时P2-NCLMO和P2-NZLMO电极的充放电GITT图及相应的Na+扩散系数。(b)根据GITT测试结果得到的反应阻力。(c)P2-NCLMO在0.1~1.0 mV s-1扫速下的CV曲线;(d)峰电流(ip)与扫速平方根(v1/2)之间的线性关系。(e)P2-NCLMO和(f)P2-NZLMO的非原位拉曼光谱。(g)还原耦合机制引起的反应动力学增强示意图。
采用恒流间歇滴定技术(GITT)评估Na+扩散系数(DNa,图4a)。计算得出P2-NCLMO的DNa值保持在10-10 cm2 s-1量级,远高于P2-NZLMO(10-15-10-10 cm2 s-1)和其他具有阴离子氧化还原化学性质的钠层氧化物正极(主要在10-12-10-11 cm2 s-1范围内)。图4b显示了由GITT结果得到的反应电阻,在整个充放电过程中,P2-NCLMO的反应电阻远低于P2-NZLMO的反应电阻。值得注意的是,在高压区域DNa值较高,反应电阻较低,表明P2-NCLMO的氧反应动力学更容易。还记录了不同扫速(0.1~1.0 mV s-1)下P2-NCLMO的CV曲线,以估计Na离子的扩散率(图4c,d)。计算得到O1、O2、O3、R1、R2、R3峰的DNa值分别为1.77 × 10-10、1.76 × 10-10、2.63 × 10-10、2.46 × 10-10、2.19 × 10-10、2.09 × 10-10 cm2 s-1。采用非原位拉曼光谱研究了TM在P2-NCLMO和P2-NZLMO中的迁移行为。约490和约590 cm-1处的峰是Na-O键的不对称拉伸(E2g)引起的,约650 cm-1处的峰是TM-O键的对称拉伸(A1g)引起的。图4e显示,E2g峰强在脱钠过程中明显降低,在钠化过程中明显增加,而P2-NCLMO的TM-O振动在充放电过程中没有明显变化,说明TM局部环境得到了很好的维持。因此,金属离子迁移在P2-NCLMO中得到有效缓解。相反,当P2-NZLMO正极充满电至4.4 V时,在≈680 cm-1处出现了一个新的尖峰(A′1g),这是由于面外阳离子迁移形成了新的TM局部环境(图4f)。放电至2.0 V时,该峰仍然存在,证实了P2-NZLMO中Zn离子从TM层向Na层的不可逆迁移。过量氧氧化后,由于Zn-O配位减弱,导致Zn2+迁移严重,Na+扩散途径受阻,随之导致Na离子扩散系数下降,反应动力学迟缓,倍率性能差,如图4g所示。
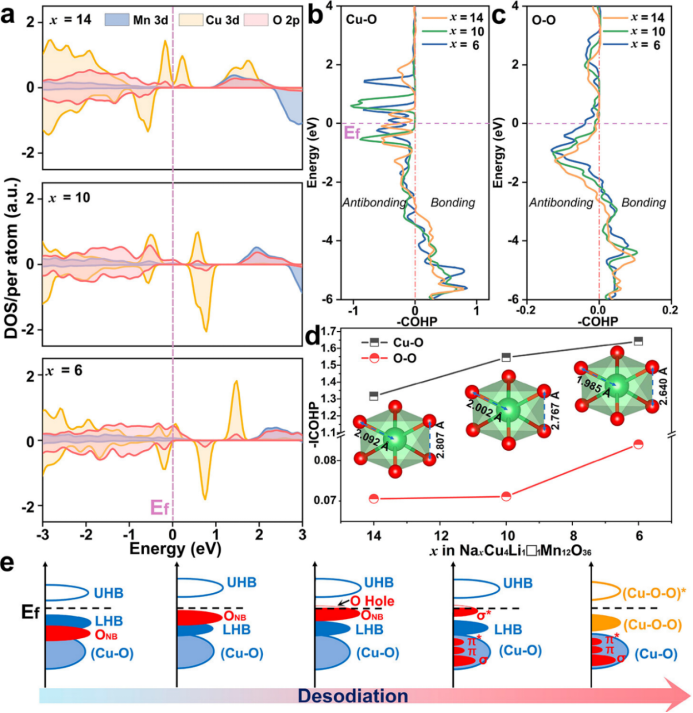
【图5】(a)NaxCu4Li1□1Mn12O36(x=14,10,6)超胞的pDOS。(b)Cu-O和(c)O-O COHP分布,(d)对应的NaxCu4Li1□1Mn12O36(x=14,10,6)的-ICOHP值。插图显示了由DFT计算得出的Cu离子(Cu-O和O-O键距离)的局部环境演变。(e)P2-NCLMO脱钠过程示意图(ONB表示非键合O 2p态;UHB和LHB分别代表上、下哈伯德能带)。
为了更好地理解还原耦合机制在稳定氧反应中的关键作用,基于密度泛函理论(DFT)对一系列NaxCu4Li1□1Mn12O36(x=14,10,6,□代表空位)超胞进行了pDOS和COHP计算。图5a显示,当x=14时,费米能级(Ef)附近的价带主要被Cu 3d轨道占据,说明在充电开始时Cu氧化还原主导了电荷补偿。当x=10时,Ef附近的上层价带主要由O 2p态组成,表明氧氧化还原在这一阶段承担了电荷补偿。在x=14~10期间,邻近Ef的Cu 3d价带明显减小,进一步表现出Cu离子的氧化。当x=6时,Ef以上的O 2p态(空穴密度)显著增加;同时,在Ef以下的Cu 3d态增加,表明在深度脱钠状态下电荷从氧转移到Cu离子(还原耦合机制)。此外,通过COHP计算研究Cu-O和O-O成键信息(图5b,c),其中正COHP值和负COHP值分别对应反键和成键状态。从Cu-O COHP谱图中可以看出,在整个脱钠过程中,Cu-O的高反键态反映了Cu-O的强相互作用,有利于氧氧化反应的稳定。然后使用积分的COHP(ICOHP)来评估Cu-O和O-O键的键合强度(图5d)。在x=14-10期间,Cu-O键的−ICOHP值的增加主要是由于Cu2+氧化为Cu3+,与Cu-O距离的缩短有很好的一致性。对于O-O键,相对较低的−ICOHP值和较长的O-O距离表明相邻氧离子之间几乎没有成键相互作用。随着Na+的进一步脱出,Cu-O和O-O键的−ICOHP值增加,键长减小,表明氧离子与Cu离子共价结合,通过还原偶联机制形成了稳定的Cu-(O-O)构型。在x=10-6期间,Cu-O键长变化较小,这是两个相反方面的综合作用:Cu离子的还原导致键长增加,而Cu-(O-O)的形成导致Cu-O距离减小。这种Cu-(O-O)相互作用可以有效地增强氧反应化学的稳定性。因此,P2-NCLMO脱钠过程中的电荷转移过程和还原耦合机理如图5e所示。
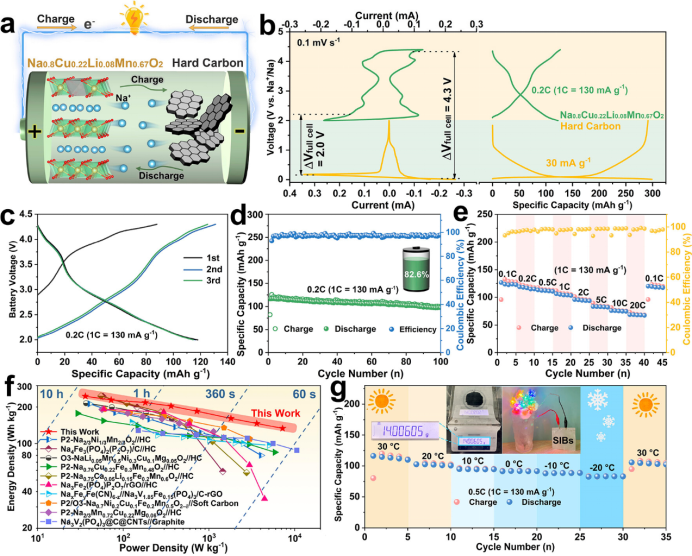
【图6】(a)P2-NCLMO//硬碳全电池示意图。(b)P2-NCLMO正极和HC负极的循环伏安图和充放电曲线。(c)恒流充放电曲线,(d)循环性能,(e)倍率性能。(f)P2-NCLMO//硬碳全电池和最近报道的钠离子全电池系统的Ragone图(基于正极和负极的总质量)。(g)全电池在不同温度下的比容量。插图显示了P2-NCLMO正极材料的批量生产和由软包电池供电的LED。
为了探索P2-NCLMO的实用性,采用P2-NCLMO正极和硬碳(HC)负极构建了钠离子电池(图6a)。根据两个配对电极的CV和充放电曲线(图6b),将全电池电压窗口设置为2.0-4.3 V,负极与正极的有效质量比固定为1:2.3。图6c、d显示了电池充满后的恒流充放电曲线和循环寿命,在0.2C下,平均工作电压为2.78 V,可逆容量为117.9 mAh g-1,根据正极和负极的总质量计算,对应的能量密度高达228.4 Wh kg-1。经过100次循环后,获得了82.6%的容量保持率。此外,全电池在0.1C和20C时的倍率性能分别为126.6 mAh g-1和68.7 mAh g-1(图6e)。图6f显示,与最近报道的其他钠离子电池相比,该电池在能量密度和功率密度上具有很大的优势。该全电池在低温下也能很好地工作(与30°C相比,-20°C的容量保持率为71.5%,图6g),并且制成的软包电池可以连续为发光二极管(LED,图6g)供电。

本工作揭示了在新型P2-Na0.8Cu0.22Li0.08Mn0.67O2正极中从O到Cu离子的不寻常电子转移的潜在还原偶联机制,以增强阴离子氧化还原反应的可逆性和动力学。Cu-O的强共价可以通过形成稳定的Cu-(O-O)相互作用抑制过量的氧氧化。因此,晶格氧损失和不可逆TM迁移同时受到抑制,这使得P2-NCLMO电极具有出色的循环稳定性(500次循环后容量保持率82%)和倍率容量(0.1C时134.1 mAh g-1, 100C时63.2 mAh g-1)。原位XRD分析表明,在Na+脱出/插入过程中,P2-NCLMO发生了固溶反应,体积变化极低,仅为1.26%。此外,GITT, CV和非原位拉曼光谱测量证实了其快速的电极动力学。通过系统的非原位XAS, XPS, XANES, EELS和EPR表征揭示了电荷转移机制,并通过DFT计算阐明了RCM稳定阴离子氧化还原化学的内在功能机制。这项研究为阴离子和阳离子协同氧化还原化学提供了深刻的见解,并为SIB的高能层状氧化物正极的开发提供了新的思路。
免责声明: 文章来自网络为作者独立观点,不代表"新能源技术与企管||程冰蕾"公众号立场。如因作品内容、版权等存在问题,请于本文刊发7日内联系"新能源技术与企管||程冰蕾"公众号进行删除或洽谈版权使用事宜
