2025年9月/10月在ECA目视检查小组成员中进行的这项调查,全面概述了各公司如何实施以及目前如何运作对注射剂产品的100%目视检查。共有115名专业人士参与了此次调查。调查结果显示,与欧盟GMP附录1(8.30 - 8.33)、USP <790> 和 <1790>、Ph. Eur. 2.9.20 和 5.17.2以及FDA在2021年12月发布的《注射剂产品可见异物检查指南(草案)》中规定的监管和药典要求基本一致。调查结果也与新版ECA目视检查指南5.0版的建议相吻合,该指南将监管期望整合到了统一的最佳实践框架中。
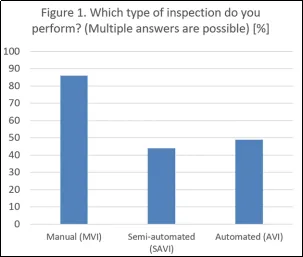
手动目视检测(MVI)仍是主要方法,86%的受访者表示采用此方法,而44%的受访者使用半自动(SAVI)系统,49%的受访者使用全自动(AVI)系统。这反映出所采用的技术种类繁多,但也证实了手动检测仍是监管参考方法,在使用AVI系统时也需要这种检测方法。
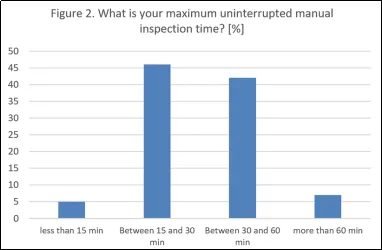
人工检查的工作条件也与GMP的期望高度契合。大多数公司称,不间断的检查时段为15至30分钟(46%)或30至60分钟(42%),其间有明确的休息时间。这些时长符合附录1(8.31)的意图,即要求频繁的休息以避免疲劳;同时也与USP <1790>关于人体工程学和操作员表现的建议相符。
同样的条款要求每年对检验员进行重新资格认定,86%的受访者都做到了这一点——这是一个极高的合规水平。只有少数受访者报告称间隔时间更长(或更短),但只要风险评估和表现趋势能证明其合理性,这种情况也是可以接受的。正如附录1和USP <1790>中所述,检验员的资格认定应包括视力检查、使用涵盖最差情况的缺陷库,以及对敏感度和误拒率的评估。
关于人工检查时是否使用放大镜,22%的受访者表示会使用放大镜,而78%的受访者表示不使用任何光学辅助工具进行检查。这一结果与预期相符,即人工检查应不使用放大镜,正如USP <790>和EP 2.9.20 所规定的,在受控照明条件下用肉眼进行目视检查。
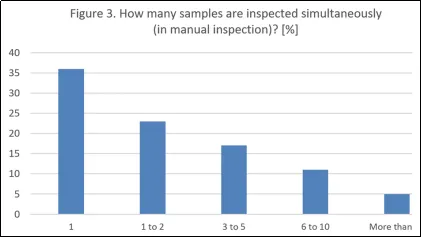
当被问及同时检查多少个药品单元时,超过半数的受访者(59%)表示他们允许同时检查1至2个单元,这也符合良好实践。然而,药典对可同时检查的样本数量没有限制。AQL数据可用于证明同时检查两个以上样本的合理性。同时检查多个容器的优势在于容易发现过量或不足的情况。
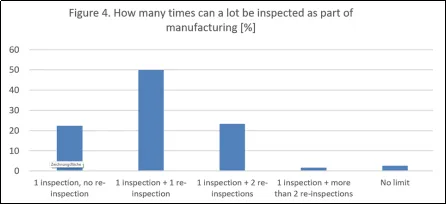
大多数公司还对作为生产流程一部分的每批产品的目视检查次数设定了明确的限制。对于“在生产过程中,一批产品可以进行多少次100%检查?”这一问题,22%的公司只允许进行一次检查且不允许复检,50%的公司允许一次复检,23%的公司允许两次复检,2%的公司允许两次以上复检,3%的公司表示没有明确的限制。这些数据非常有趣。不允许任何复检(22%)的规定非常严格。73%的公司允许在生产过程中进行一次或两次复检,这与现行法规(FDA倾向于只允许一次复检)相符。然而,5%的公司允许进行两次以上复检或对100%目视检查的次数没有限制。这可能会通过将目视检查描述为纯粹的生产步骤而非测试来加以辩解。然而,如果复检次数经常很高,那么在GMP检查中可能需要对先前生产流程的有效性作出解释。
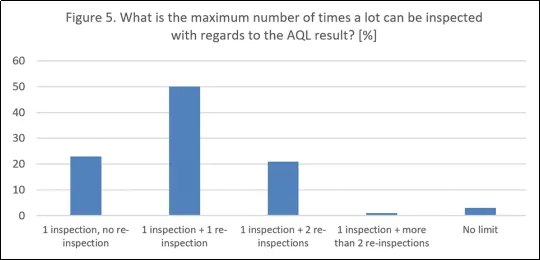
大多数公司(50%)允许在AQL检验不合格后对一批产品进行一次复检,21%的公司允许进行两次复检,而一小部分(3%)公司表示没有明确的限制。这种无限制的复检做法存在合规风险,因为附录1要求对异常缺陷水平进行调查和趋势分析,而USP仅在经过验证和控制的条件下才允许进行第二次检验。新的ECA指南 5.0 提出了一种在AQL检验中发现颗粒时应遵循的程序,建议只有在完成书面根本原因分析、采取纠正措施并收紧AQL接收标准后,才进行第二次100%检验。
几乎所有参与者(97%)在完成100%检查步骤后都会进行可接受质量水平(AQL)测试,这正是USP中所描述的方法。进行AQL测试的责任主要由质量保证/质量控制部门承担(占 56%),而42%的人表示生产人员将AQL测试作为生产过程的一部分来进行。这种分布情况表明,更多的企业将AQL视为质量控制活动,而非生产过程中的检查环节。
关于颗粒分类问题,52%的受访者将可见颗粒视为关键缺陷,44%认为其为重大缺陷,仅有 4%认为其为轻微缺陷。这种分布情况表明,大多数公司采取保守的处理方式,将可见颗粒视为关键缺陷。USP将可见颗粒描述为可能对患者安全构成威胁的缺陷,并且尽管药典也建议将其作为批次级验收决策中的重大缺陷(对于非外在颗粒而言),但此次调查显示,内部的分类实践比药典规定的最低标准更为严格。
当被问及自动化系统的验证标准时,63%的受访者认为其接受标准应基于性能不低于或优于人工检查的水平;而26%的受访者则针对不同缺陷的严重程度设定固定的检测率目标。这两种方法均得到了USP的支持,USP将人工检查视为参考标准,并允许采用诸如检测概率(PoD)等定量性能指标。因此,与人工检查结果的比较仍然是所谓的“黄金标准”或参考标准。
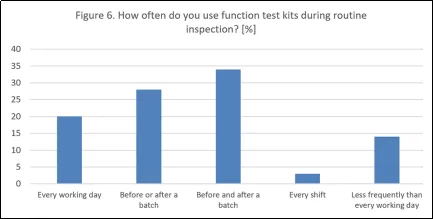
在常规检查中使用功能测试套件这一做法也体现了成熟的操作规范:34%的受访者会在每批产品生产前后进行测试,28%的受访者会在生产前后进行测试,还有20%的受访者会在每个工作日进行测试。这些结果符合附录1(8.32)的要求,该附件要求在系统启动前以及定期对自动化系统进行验证测试。它们也与USP和ECA 5.0中的建议相一致,这两份文件都强调了对自动化检测设备进行持续性能验证的重要性。
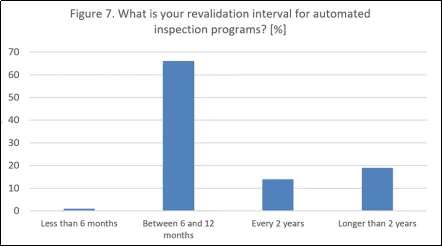
还询问了自动化视觉检测(AVI)系统再验证的频率。结果显示,大多数公司会在6至12个月内进行重新验证(占66%),这相当于1个年度周期,与当前的监管要求高度一致。仅有1%的公司报告重新验证周期少于6个月,这种情况通常出现在流程或其他方面发生变化需要提前审查的情况下。一小部分公司每2年进行一次重新验证(占14%),而19%的公司表示周期超过2年。因此,这项调查表明,三分之二的公司遵循每年或更短周期的重新验证频率。
目前,人工智能尚未在视觉检测中得到广泛应用;仅有1%的受访者目前采用了基于人工智能的算法,而27%的受访者计划在不久的将来这样做。这种谨慎的采用方式与当前的监管环境相一致。尽管目前没有药典或GMP规定明确提及人工智能,但其使用遵循与任何自动分类器相同的验证原则:与人工参考的等效性、数据集的代表性、持续监测偏差情况以及有记录的变更控制。ECA指南5.0和USP都强调,任何新型技术都必须证明具有同等或更高的检测能力,并保持过程控制的稳定性。
总体而言,该调查表明行业与主要的监管和药典要求高度契合。人工检查仍作为参考方法发挥着核心作用,而自动化系统则越来越多地通过验证达到与之相当甚至更优的性能水平。AQL测试和趋势分析已得到广泛应用,大多数公司都遵循公认的最佳实践来进行功能挑战测试和操作人员资格认证。仍需改进的领域包括在AQL失效后重新检查限值的一致定义,以及通过缺陷类别对自动化系统性能进行定量评估。通过解决这些问题,公司能够确保完全符合欧盟GMP附录 1、USP以及EP 2.9.20和5.17.2的要求,并遵循在ECA目视检查指南第 5.0 版中整合的实用建议。
原文来自GMP Journal网站,点击“阅读原文”可查看原文。
更多目视检查相关的文章见:
#ECA #目视检查 #AQL